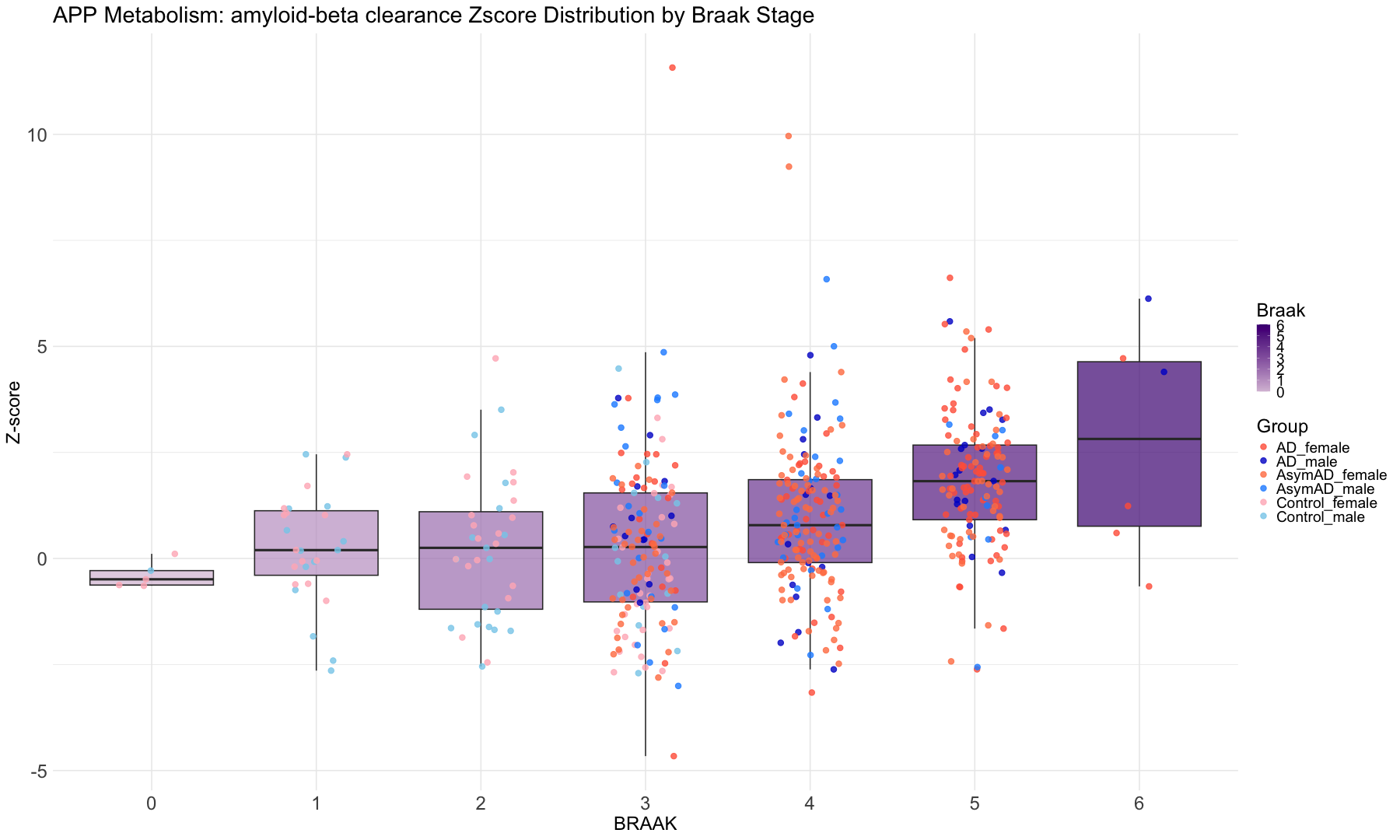
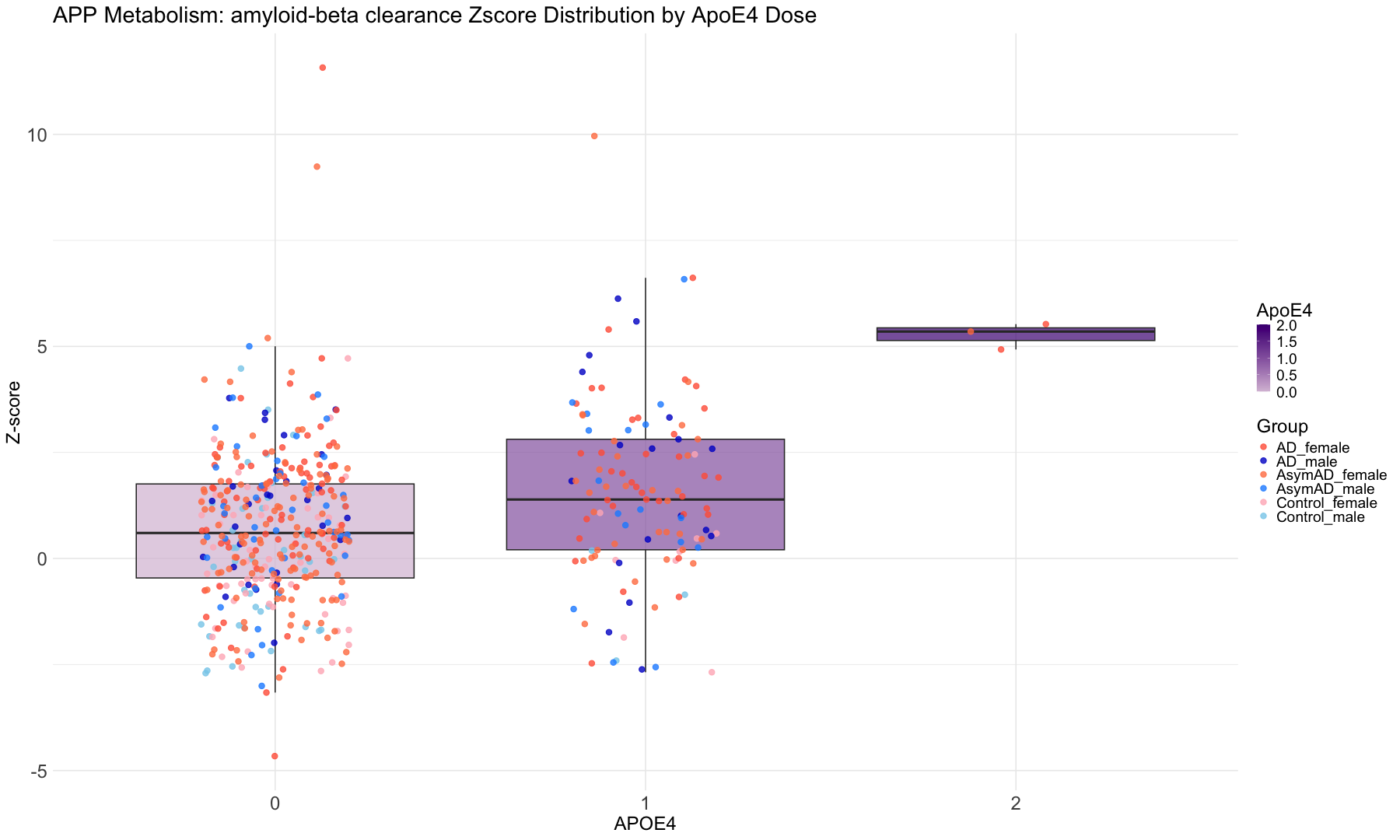
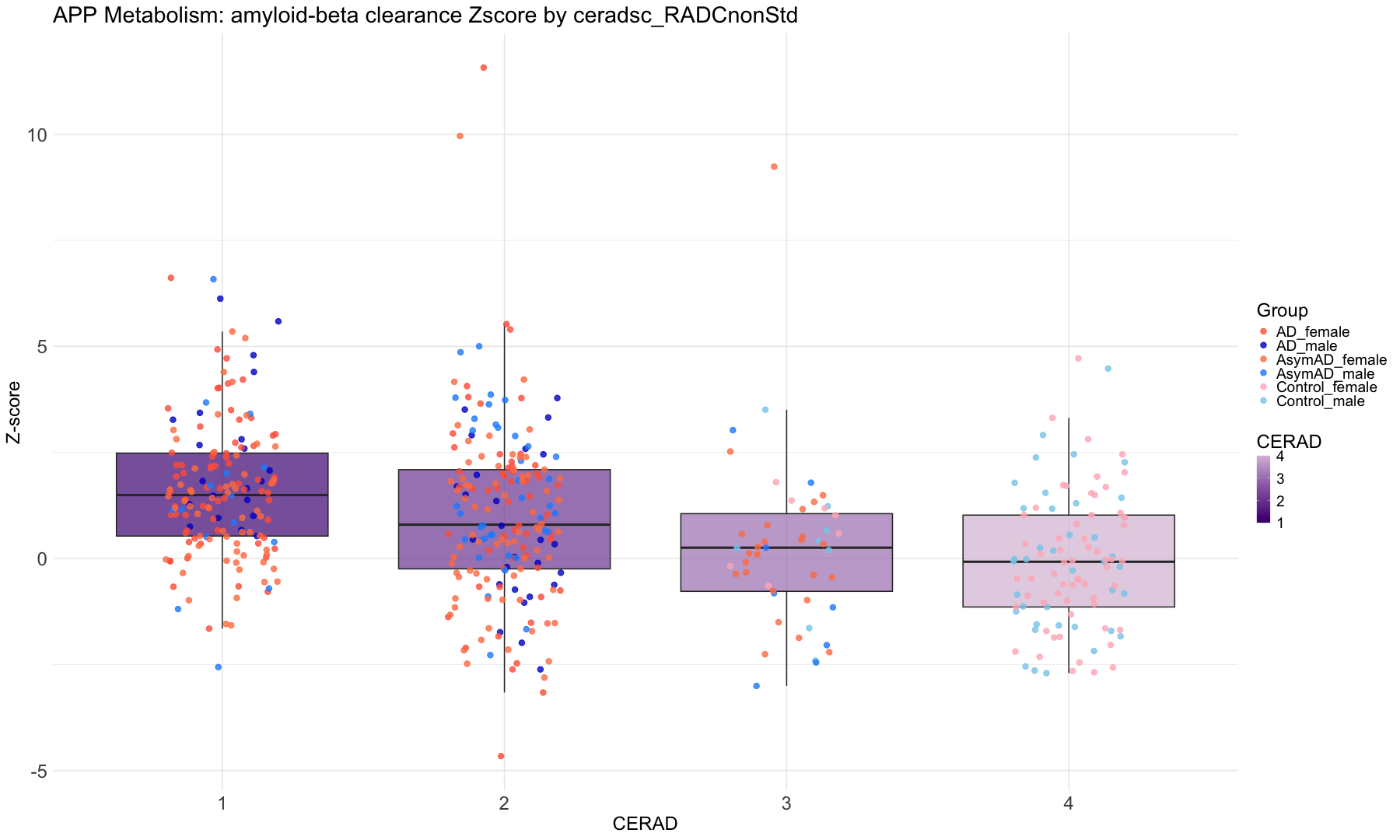
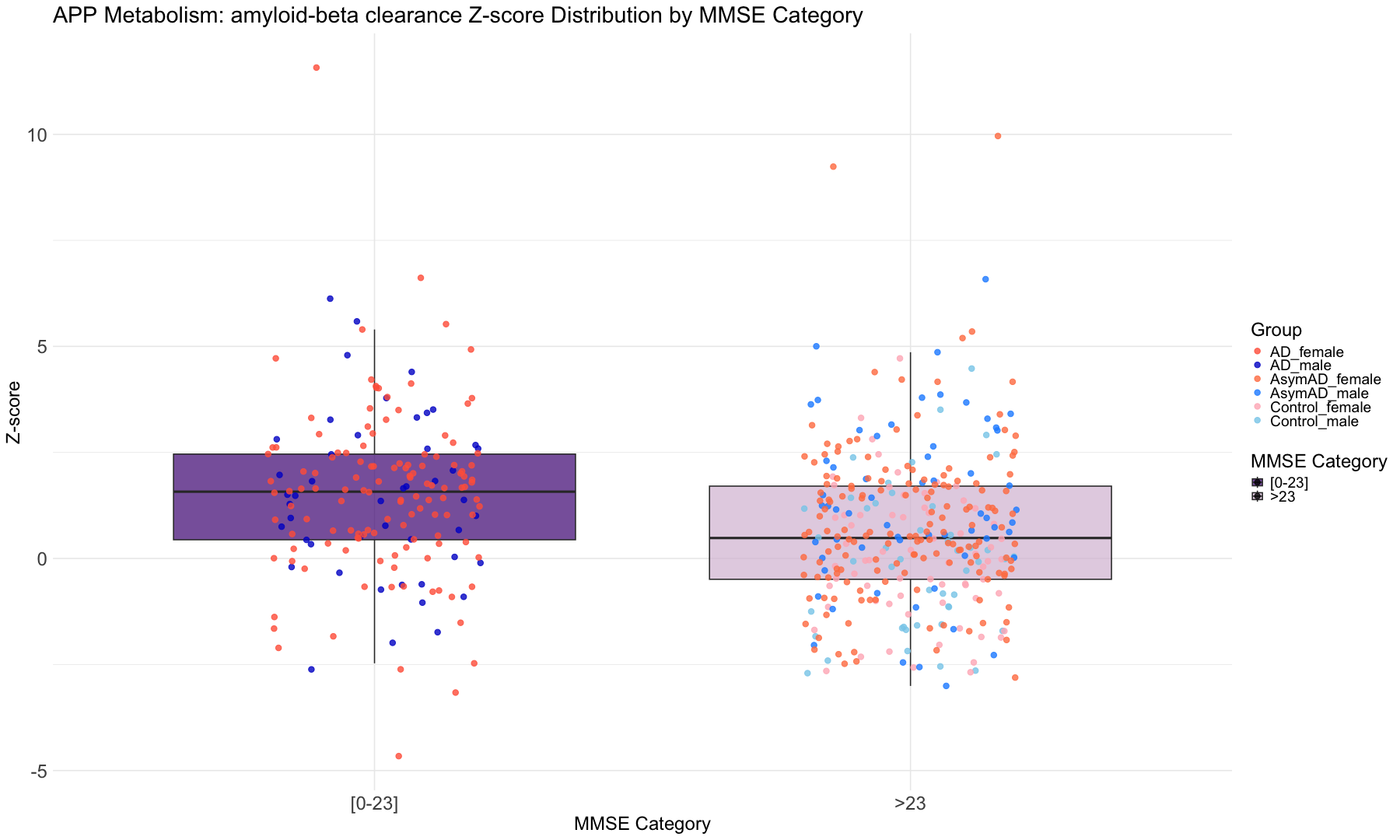
library(dplyr)
library(ggplot2)
library(stringr)
library(ggnewscale)
# Open the merged clinical parameter correlation table for the sigificant subdomain in each catagory (P value < 0.05)
data <- read.csv("clinical_parameter_correlation_all_FDR_062325.csv", sep =",", header = TRUE, stringsAsFactors = FALSE)
#data <- read.csv("clinical_parameter_correlation_spearman.csv", sep =",", header = TRUE, stringsAsFactors = FALSE)
head(data)
# Define the color code for each Biodomain
custom_colors <- c("Apoptosis" = "#673399", "APP Metabolism" = "#fe6500", "Autophagy" = "#9931fd", "Cell Cycle" = "#18857f", "DNA Repair" = "#f451ad", "Endolysosome" = "#3466cc", "Epigenetic" = "#cb3233", "Immune Response" = "#9ccdcc", "Lipid Metabolism" = "#989898", "Metal Binding and Homeostasis" = "#4b0d20", "Mitochondrial Metabolism" = "#97cb98", "Myelination" = "#996735", "Oxidative Stress" = "#ffcd66", "Proteostasis" = "#c8b269", "RNA Spliceosome" = "#0c9aff", "Structural Stabilization" = "#ff9a9a", "Synapse" = "#329a33", "Tau Homeostasis" = "#cb97cb", "Vasculature" = "#cecd02", "none" = "#7f7f7f")
# Wrap long Biodomain_Subdomain names
#data <- data %>%
#mutate(Biodomain_Subdomain_wrapped = str_wrap(Biodomain_Subdomain, width = 40))
# Truncate long Biodomain_Subdomain names to 40 characters
#data <- data %>%
#mutate(Biodomain_Subdomain_short = str_trunc(Biodomain_Subdomain, width = 40))
# Step 1: Rename only selected Metadata values, leave others unchanged
data$Metadata <- recode(data$Metadata,
"ApoE4.Dose" = "APOE4",
"braaksc" = "BRAAK",
"ceradsc_RADCnonStd" = "CERAD",
"cts_mmse30_lv"= "MMSE"
)
# Step 2: Reorder Metadata as factor with desired order
data$Metadata <- factor(data$Metadata, levels = c("APOE4", "CERAD", "BRAAK", "MMSE"))
# Step 3: Identify Biodomain_Subdomain values that appear in more than one Metadata
#common_subdomains <- data %>%
#group_by(Biodomain_Subdomain) %>%
#summarise(n_metadata = n_distinct(Metadata)) %>%
#filter(n_metadata > 1) %>%
#pull(Biodomain_Subdomain)
# Step 4: Add a flag to the original data
#data <- data %>%
#mutate(is_common = Biodomain_Subdomain %in% common_subdomains)
# Step 5:Order data by Biodomain and Biodomain_Subdomain
data <- data %>%
arrange(Biodomain, Biodomain_Subdomain)
# Step 6:Create factor with correct order
data$Biodomain_Subdomain <- factor(data$Biodomain_Subdomain, levels = unique(data$Biodomain_Subdomain))
# Step 7:Determine y positions for rectangles
biodomain_bounds <- data %>%
distinct(Biodomain, Biodomain_Subdomain) %>%
group_by(Biodomain) %>%
summarise(
ymin = min(as.numeric(Biodomain_Subdomain)) - 0.5,
ymax = max(as.numeric(Biodomain_Subdomain)) + 0.5
)
# Plot
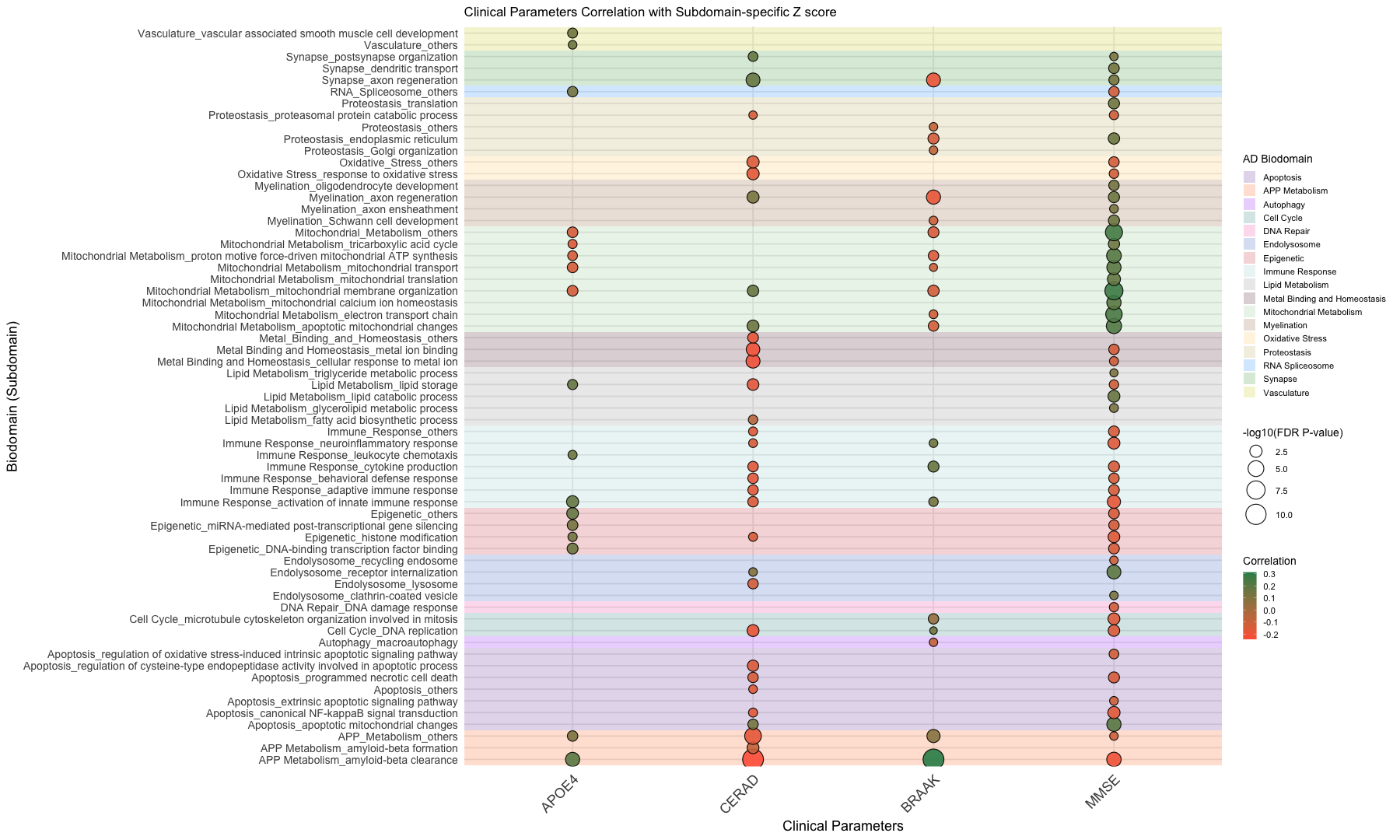
ggplot(data, aes(x = Metadata, y = Biodomain_Subdomain)) +
# Draw background boxes per Biodomain group
geom_rect(data = biodomain_bounds,
aes(xmin = -Inf, xmax = Inf, ymin = ymin, ymax = ymax, fill = Biodomain),
inherit.aes = FALSE, alpha = 0.20) +
scale_fill_manual(values = custom_colors, name = "AD Biodomain") +
# Reset fill scale for correlation bubble color
new_scale_fill() +
# Bubble plot layer
geom_point(aes(fill = correlation, size = nl10_FDR_p_value),
shape = 21, color = "black", alpha = 0.9) +
scale_fill_gradient(low = "tomato", high = "seagreen", name = "Correlation") +
scale_size(range = c(4, 12), name = "-log10(FDR P-value)") +
# Box outline overlay for common points
#geom_point(
#data = subset(data, is_common),
#aes(x = Metadata, y = Biodomain_Subdomain),
#shape = 0, # square outline
#stroke = 1.5, # Line thickness
#color = "black", # Outline color
#fill = NA, # Transparent fill to create a "box" effect
#size = 8 # Match or slightly larger than base point size
#) +
# Labels and theme
labs(
title = "Clinical Parameters Correlation with Subdomain-specific Z score",
x = "Clinical Parameters",
y = "Biodomain (Subdomain)"
) +
theme_minimal(base_size = 18) +
theme(
text = element_text(size = 14),
axis.text.x = element_text(size = 18, hjust = 1, angle = 45),
axis.text.y = element_text(size = 14),
axis.title = element_text(size = 18),
strip.text = element_text(size = 24)
)