##
## Attaching package: 'gplots'
## The following object is masked from 'package:stats':
##
## lowess
## Warning: package 'ggplot2' was built under R version 4.5.2
library(reshape2)
library(RColorBrewer)
library(dplyr)
##
## Attaching package: 'dplyr'
## The following objects are masked from 'package:stats':
##
## filter, lag
## The following objects are masked from 'package:base':
##
## intersect, setdiff, setequal, union
## Loading required package: viridisLite
library(ggrepel)
library(corrplot)
##
## Attaching package: 'plotly'
## The following object is masked from 'package:ggplot2':
##
## last_plot
## The following object is masked from 'package:stats':
##
## filter
## The following object is masked from 'package:graphics':
##
## layout
library(patchwork)
library(stringr)
# Open subdomains comparison file"
#data_subdomain <- read.csv("comarative_subdomain_results.csv", sep =",", header = TRUE, stringsAsFactors = FALSE)
data_subdomain <- read.csv("comarative_subdomain_results_050525.csv", sep =",", header = TRUE, stringsAsFactors = FALSE)
# Truncate long Biodomain_Subdomain names to 40 characters
data_subdomain <- data_subdomain %>%
mutate(Biodomain_Subdomain = str_trunc(Biodomain_Subdomain, width = 40))
# Removing the 1st and 2nd columns
data_subdomain_new_UP <- data_subdomain[, -c(1, 2, 3, 4, 7, 9, 11, 13)]
head(data_subdomain_new_UP)
## Biodomain_Subdomain UP_ratio_AD_female UP_ratio_AD_male
## 1 APP Metabolism_amyloid-beta clearance 0.1015625 0.08510638
## 2 APP Metabolism_amyloid-beta formation 0.1875000 0.00000000
## 3 APP Metabolism_amyloid precursor prot... 0.0000000 0.10638298
## 4 APP Metabolism_amyloid precursor prot... 0.0234375 0.04255319
## 5 APP Metabolism_other 0.0234375 0.04255319
## 6 Apoptosis_apoptotic mitochondrial cha... 0.0390625 0.00000000
## UP_ratio_AsymAD_female UP_ratio_AsymAD_male
## 1 0.050314465 0.04918033
## 2 0.157232704 0.03278689
## 3 0.006289308 0.08196721
## 4 0.012578616 0.01639344
## 5 0.031446541 0.01639344
## 6 0.050314465 0.00000000
data_subdomain_new_DOWN <- data_subdomain[, -c(1, 2, 3, 4, 6, 8, 10, 12)]
head(data_subdomain_new_DOWN)
## Biodomain_Subdomain DOWN_ratio_AD_female
## 1 APP Metabolism_amyloid-beta clearance 0.0234375
## 2 APP Metabolism_amyloid-beta formation 0.0234375
## 3 APP Metabolism_amyloid precursor prot... 0.0781250
## 4 APP Metabolism_amyloid precursor prot... 0.0078125
## 5 APP Metabolism_other 0.0156250
## 6 Apoptosis_apoptotic mitochondrial cha... 0.0546875
## DOWN_ratio_AD_male DOWN_ratio_AsymAD_female DOWN_ratio_AsymAD_male
## 1 0.00000000 0.006289308 0.01639344
## 2 0.06382979 0.006289308 0.06557377
## 3 0.06382979 0.056603774 0.09836066
## 4 0.04255319 0.006289308 0.00000000
## 5 0.06382979 0.025157233 0.03278689
## 6 0.12765957 0.012578616 0.03278689
data_matrix_subdomain_UP <- data_subdomain_new_UP[, -1]
# Reorder the columns
data_matrix_subdomain_UP <- data_matrix_subdomain_UP[, c(
"UP_ratio_AD_female",
"UP_ratio_AsymAD_female",
"UP_ratio_AD_male",
"UP_ratio_AsymAD_male"
)]
data_matrix_subdomain_DOWN <- data_subdomain_new_DOWN[, -1]
# Reorder the columns
data_matrix_subdomain_DOWN <- data_matrix_subdomain_DOWN[, c(
"DOWN_ratio_AD_female",
"DOWN_ratio_AsymAD_female",
"DOWN_ratio_AD_male",
"DOWN_ratio_AsymAD_male"
)]
# Replace NA values with 0 (or any other specific value)
data_matrix_subdomain_UP[is.na(data_matrix_subdomain_UP)] <- 0
data_matrix_subdomain_DOWN[is.na(data_matrix_subdomain_DOWN)] <- 0
# Set up the color scale using RColorBrewer
color_scale <- colorRampPalette(brewer.pal(9, "RdYlGn"))
# Assign custom column names
colnames(data_matrix_subdomain_UP) <- c("AD_Female", "AsymAD_Female", "AD_Male", "AsymAD_Male")
# Assign custom column names
colnames(data_matrix_subdomain_DOWN) <- c("AD_Female", "AsymAD_Female", "AD_Male", "AsymAD_Male")
# Set up the color scale using RColorBrewer
color_scale <- colorRampPalette(brewer.pal(9, "RdYlGn"))
color_scale1 <- colorRampPalette(c("white", "lightblue", "blue", "darkblue"))
color_scale2 <- colorRampPalette(brewer.pal(9, "RdGy"))
color_scale3 <- colorRampPalette(brewer.pal(9, "Greens"))
color_scale4 <- colorRampPalette(brewer.pal(9, "Blues"))
color_scale5 <- colorRampPalette(brewer.pal(9, "Purples"))
color_scale6 <- colorRampPalette(brewer.pal(9, "Oranges"))
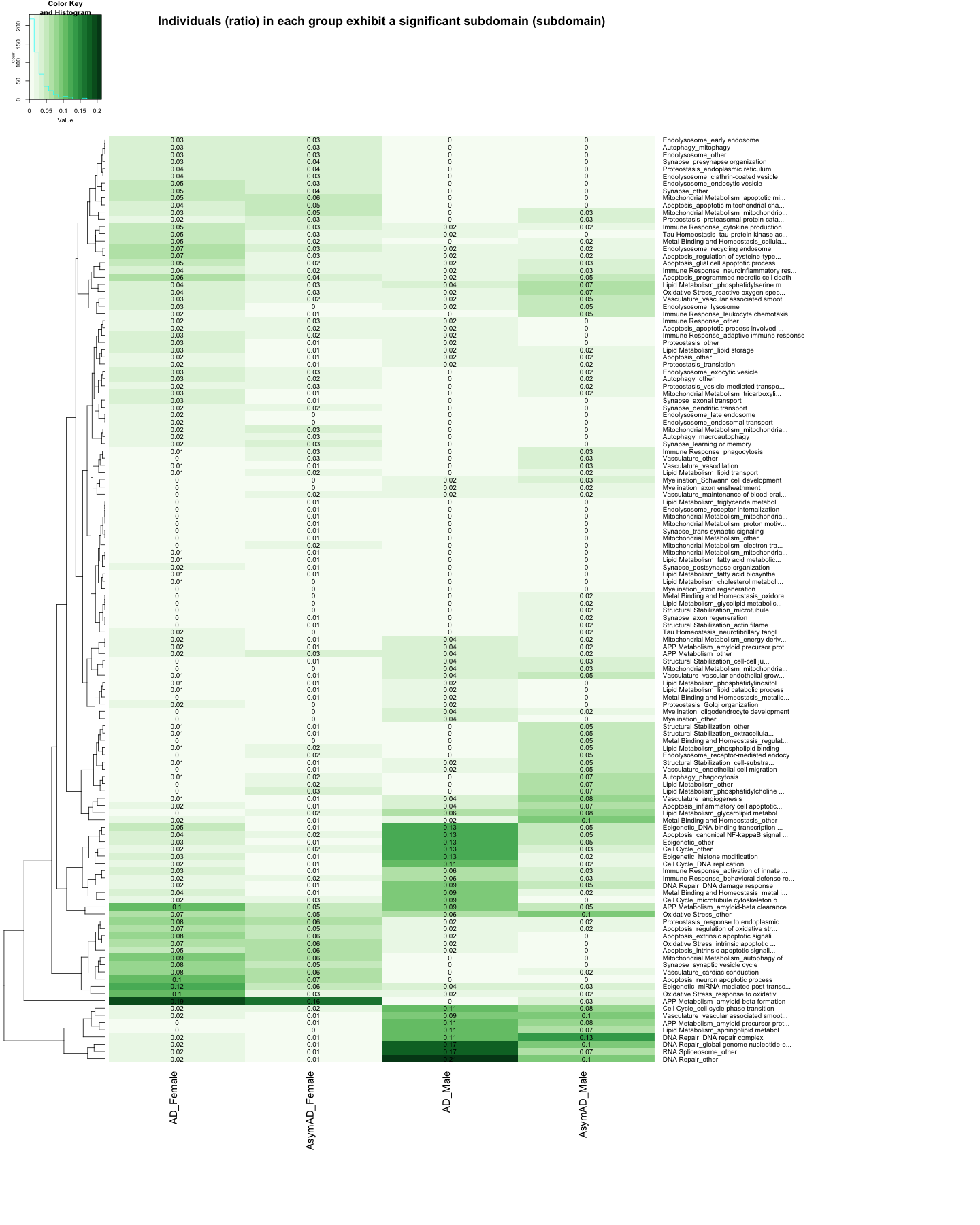
heatmap.2(
as.matrix(data_matrix_subdomain_UP),
Rowv = TRUE,
Colv = FALSE,
col = color_scale3,
scale = "none", # Scale rows (genes) to Z-scores
trace = "none", # Turn off row and column annotations
margins = c(20, 40), # Set margins for row and column labels
key = TRUE, # Include a color key
keysize = .5, # Size of the color key
cexCol = 1.5, # Set column label size
cexRow = 1, # Set row label size
cellnote=round(data_matrix_subdomain_UP, 2),
notecol="black",
labRow = data_subdomain$Biodomain_Subdomain, # Row labels (subdomain name)
dendrogram = "row", # Show both row and column dendrograms
main="Individuals (ratio) in each group exhibit a significant subdomain (subdomain)"
)
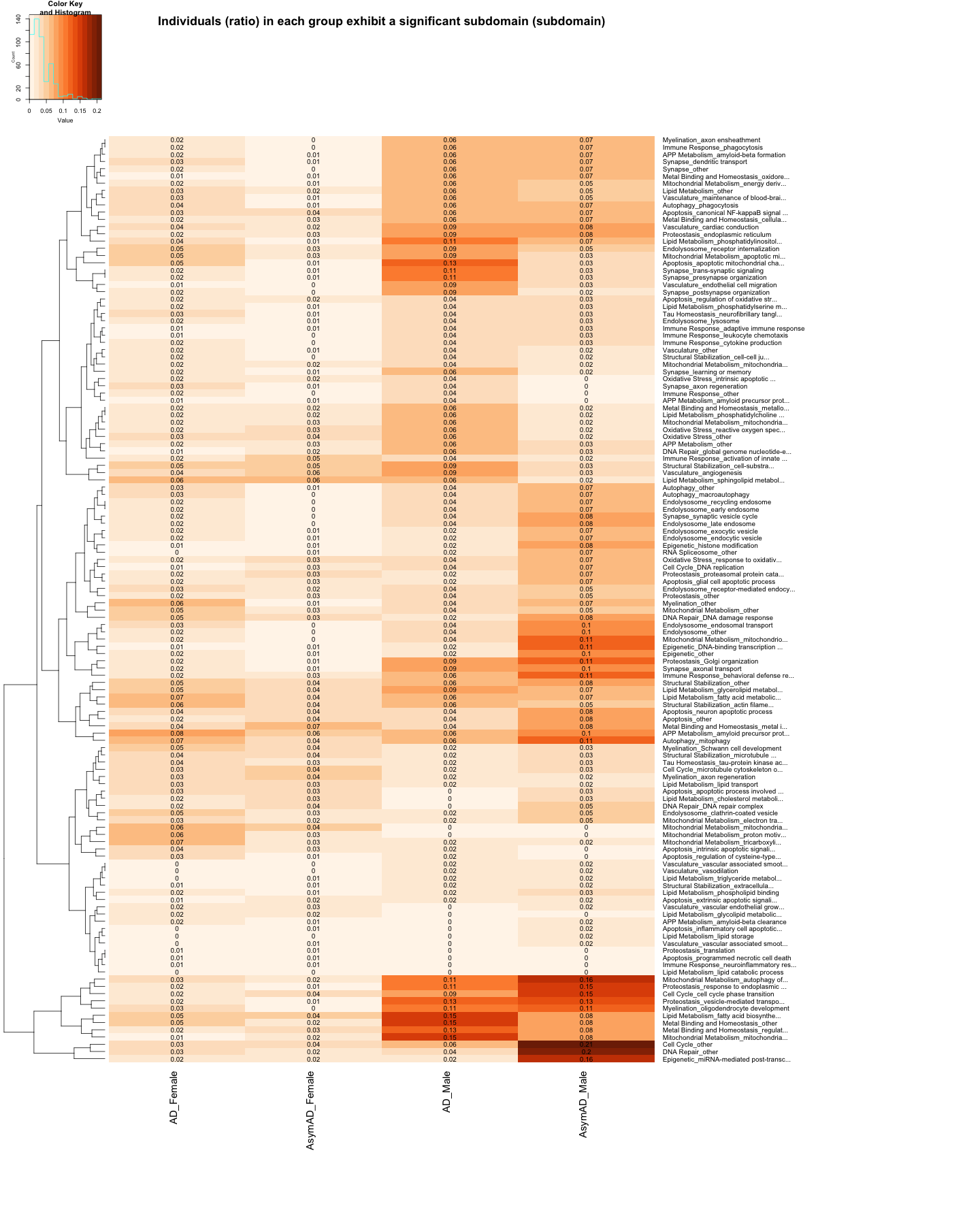
heatmap.2(
as.matrix(data_matrix_subdomain_DOWN),
Rowv = TRUE,
Colv = FALSE,
col = color_scale6,
scale = "none", # Scale rows (genes) to Z-scores
trace = "none", # Turn off row and column annotations
margins = c(20, 40), # Set margins for row and column labels
key = TRUE, # Include a color key
keysize = .5, # Size of the color key
cexCol = 1.5, # Set column label size
cexRow = 1, # Set row label size
cellnote=round(data_matrix_subdomain_DOWN, 2),
notecol="black",
labRow = data_subdomain$Biodomain_Subdomain, # Row labels (subdomain name)
dendrogram = "row", # Show both row and column dendrograms
main="Individuals (ratio) in each group exhibit a significant subdomain (subdomain)"
)
########################################################################################################################################################################################################
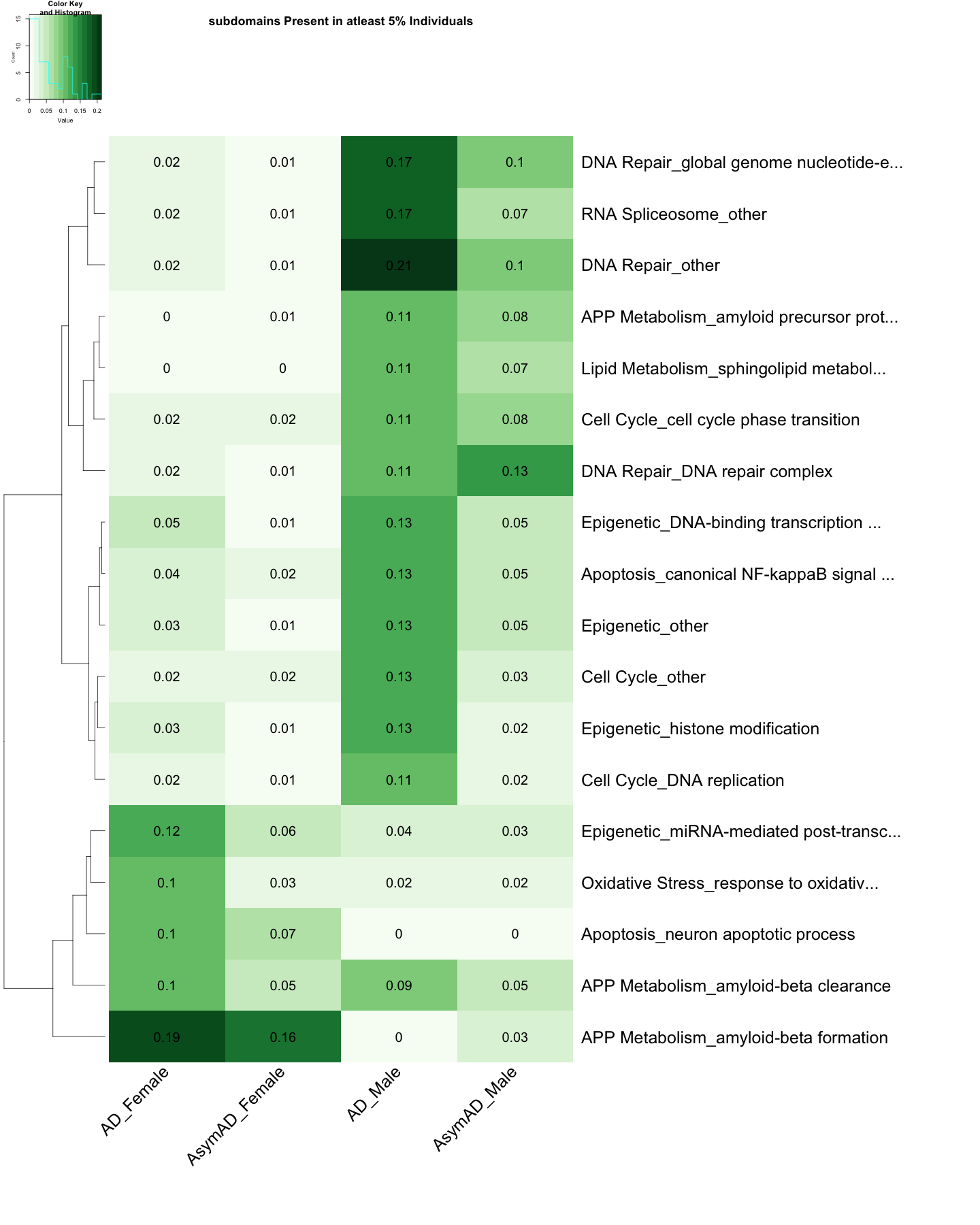
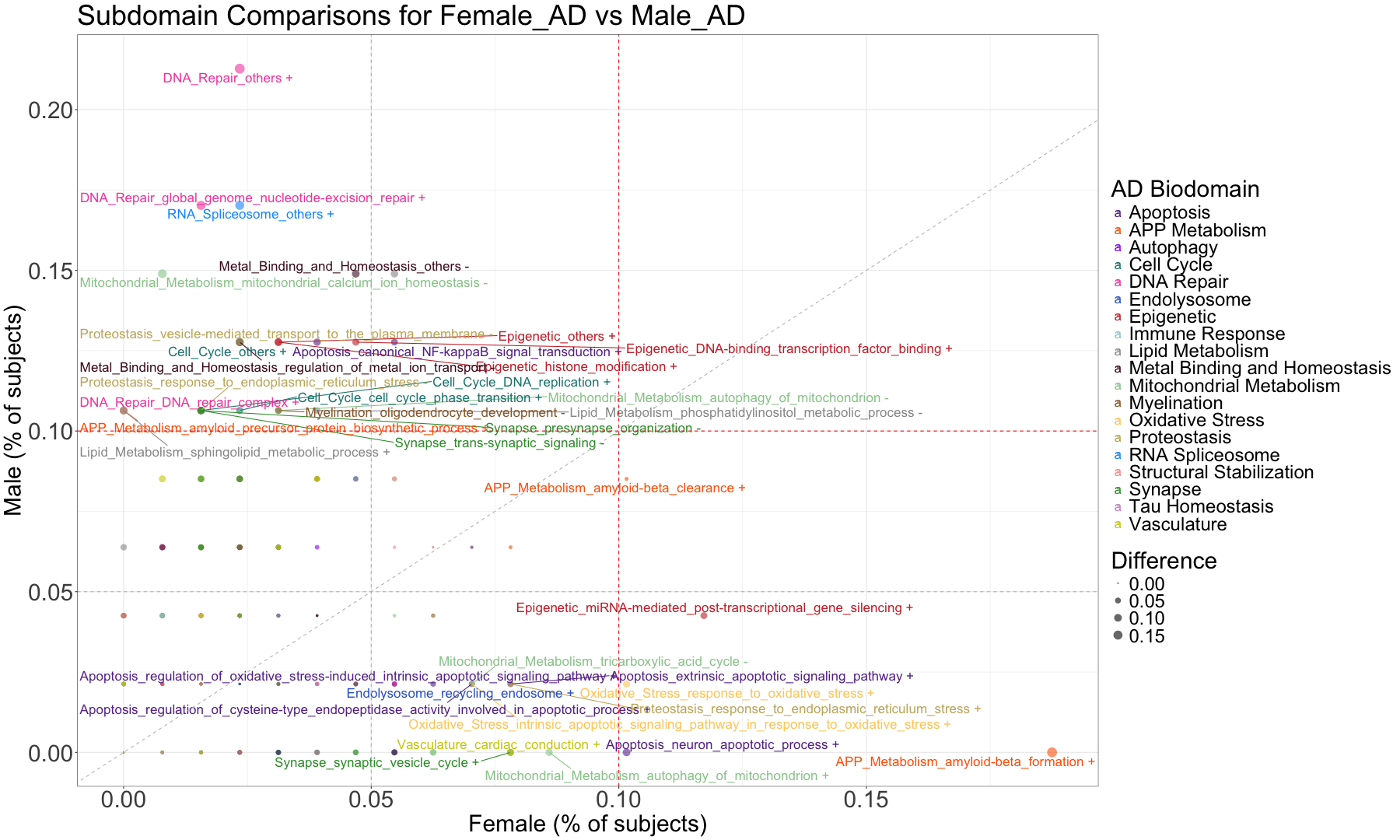
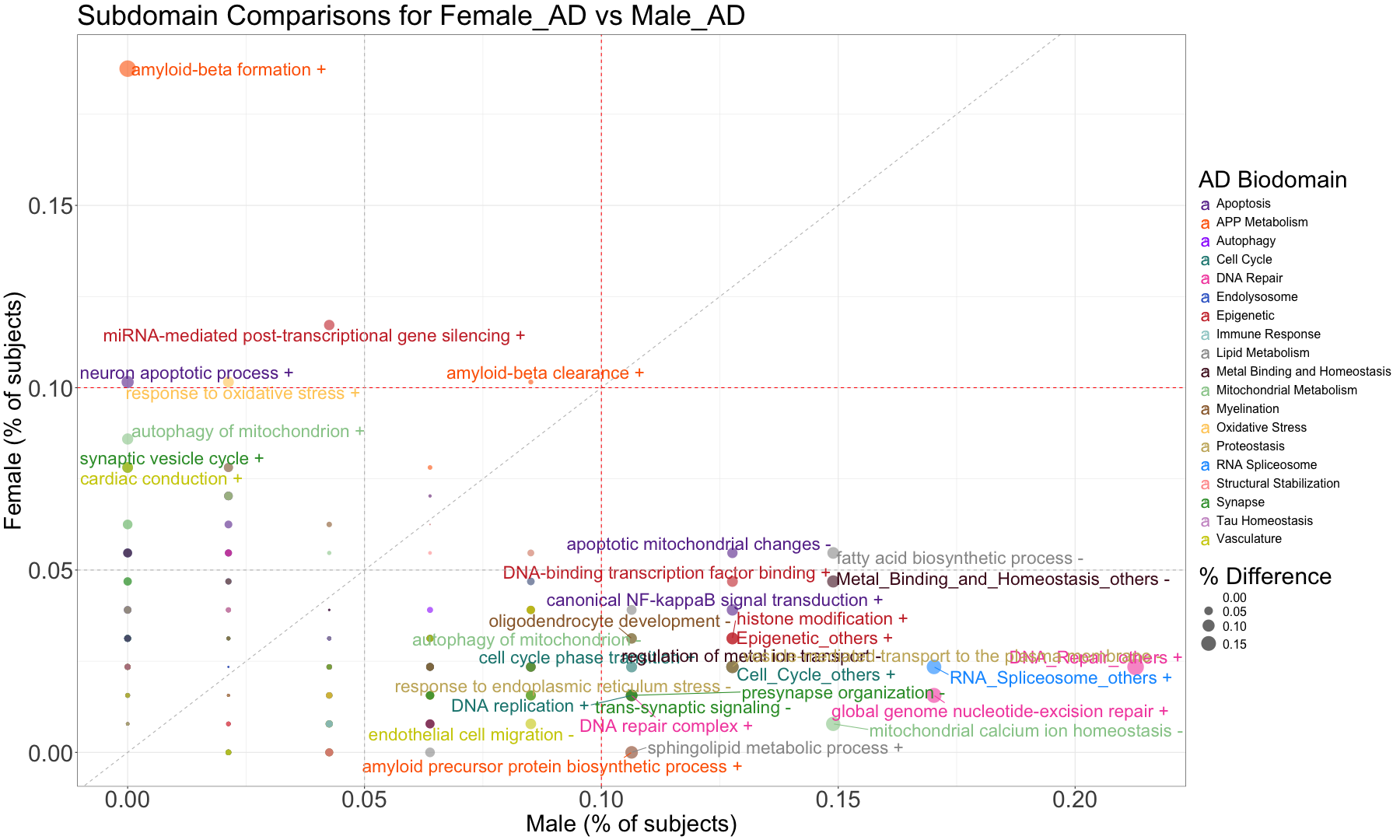
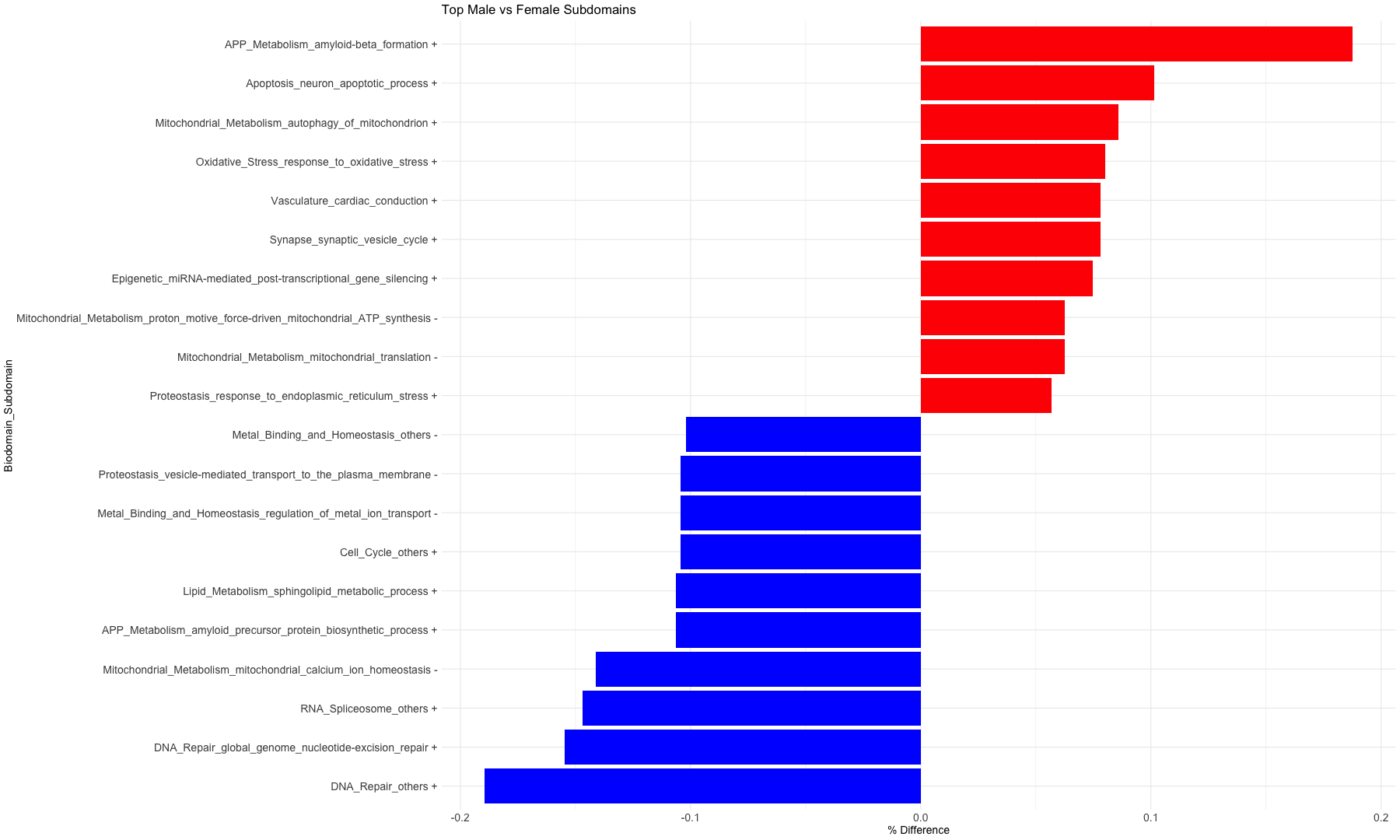
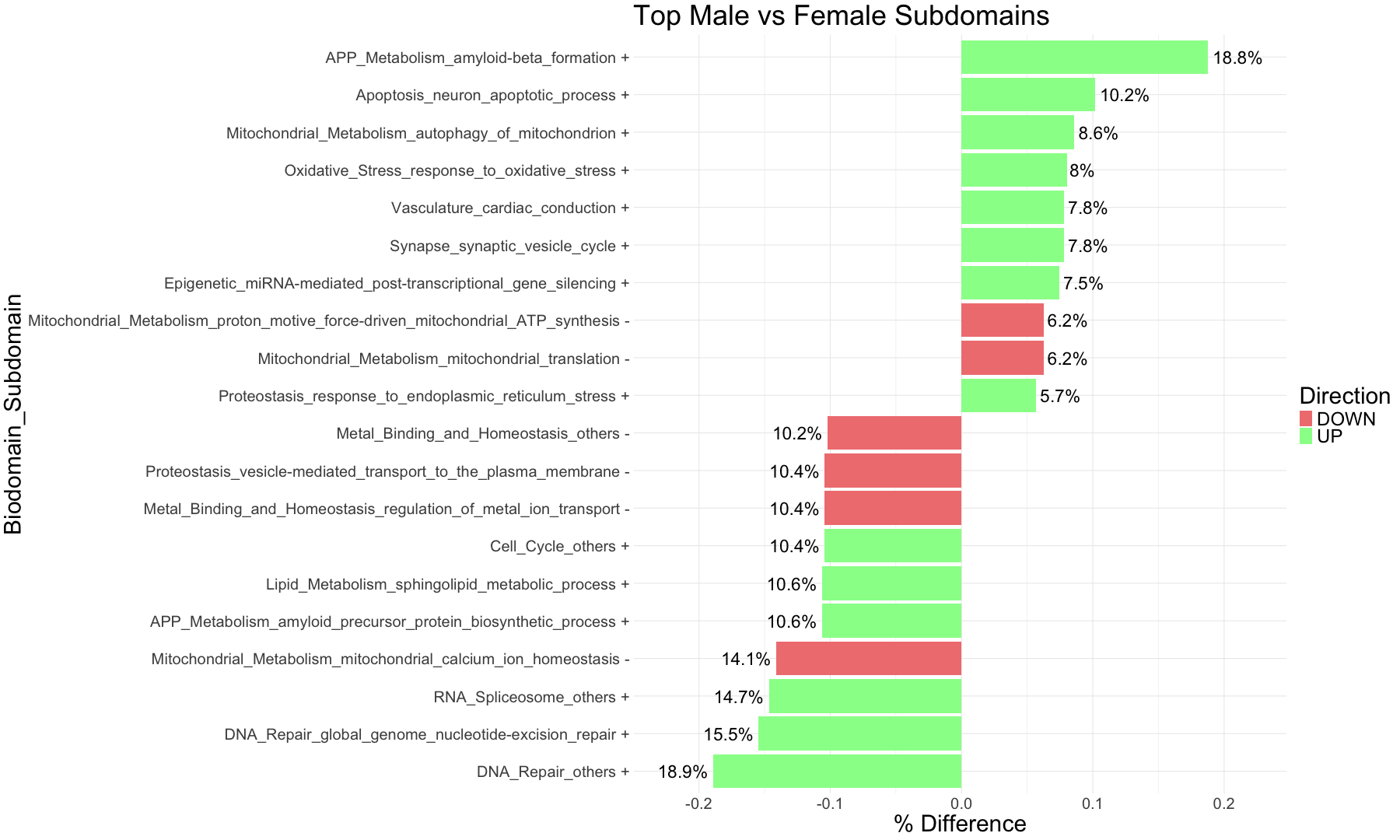
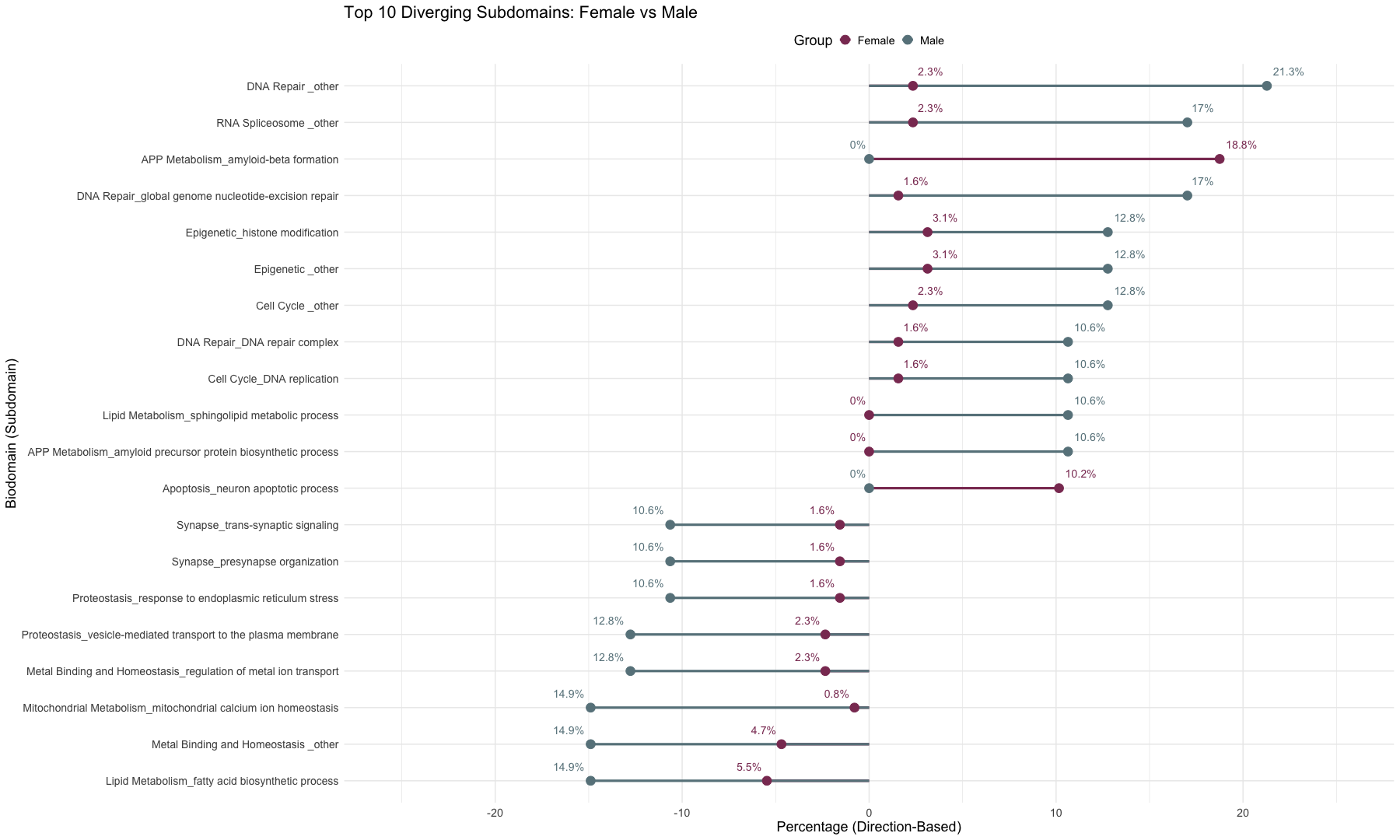
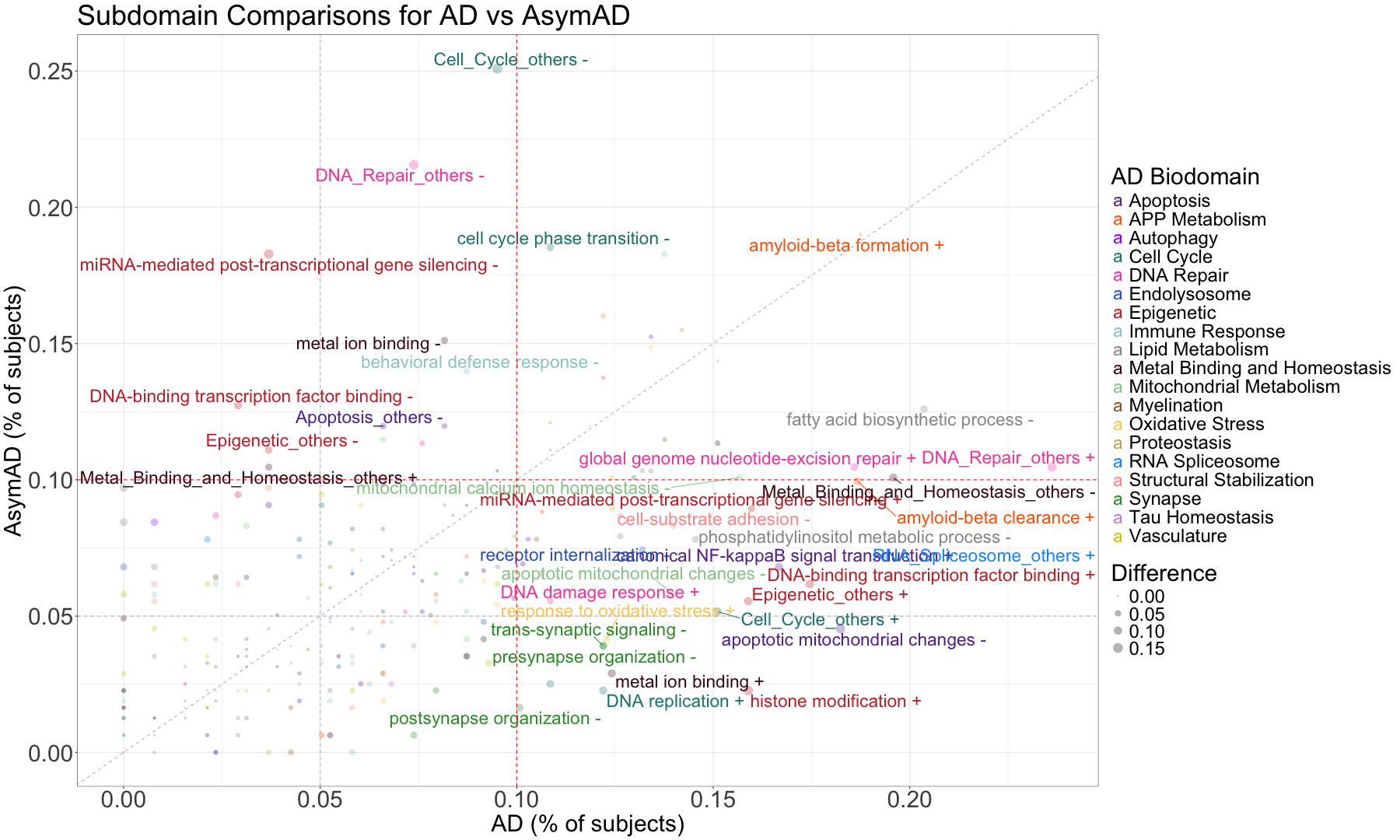
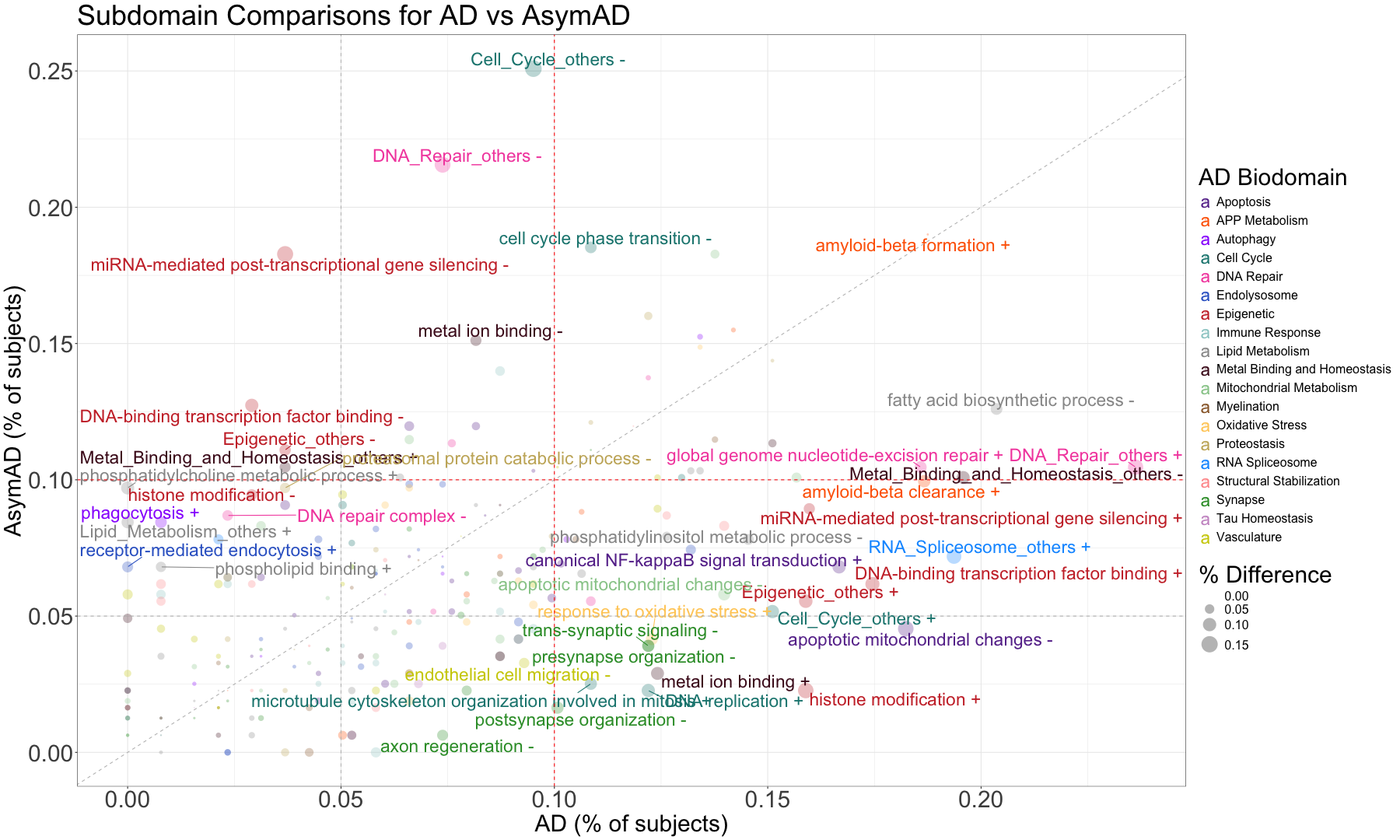
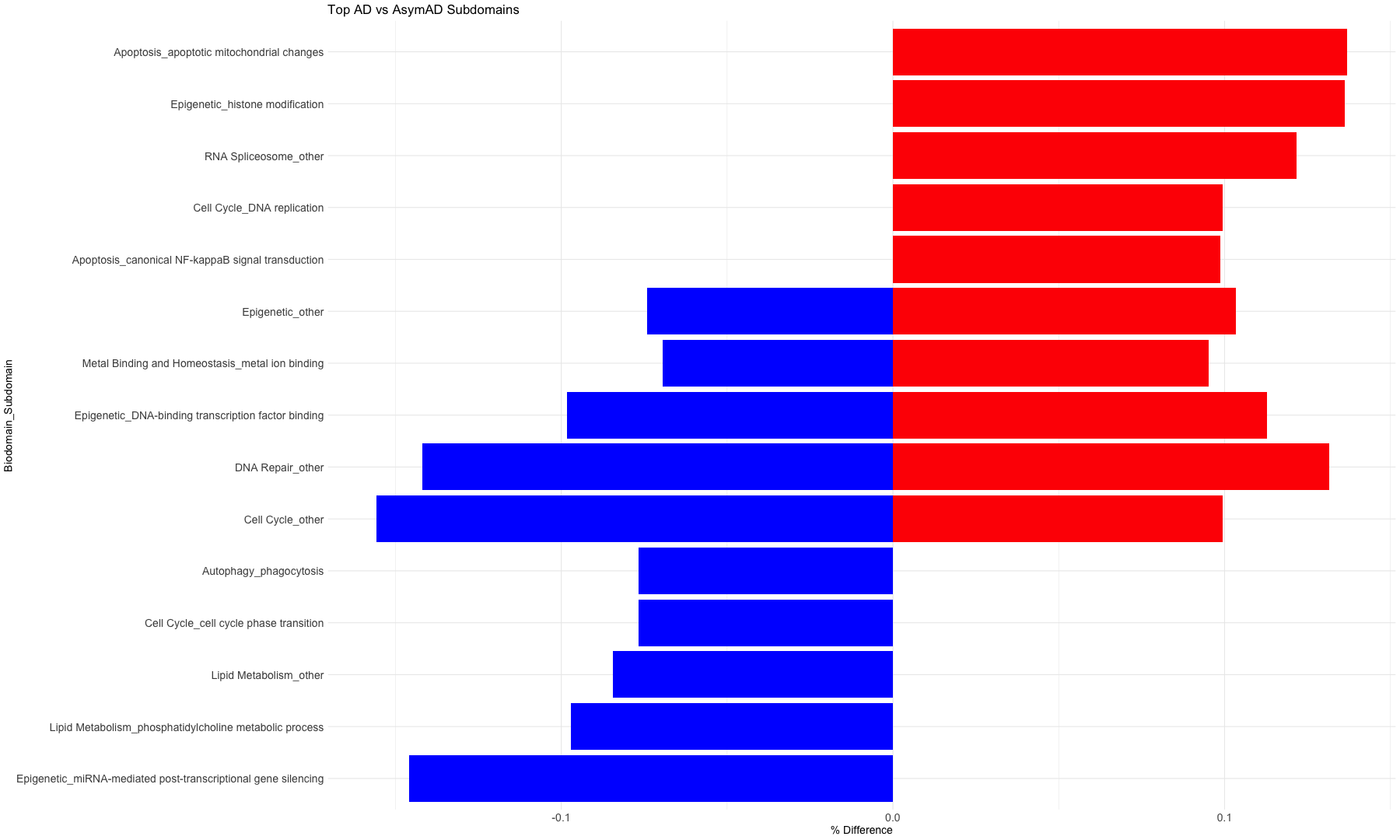
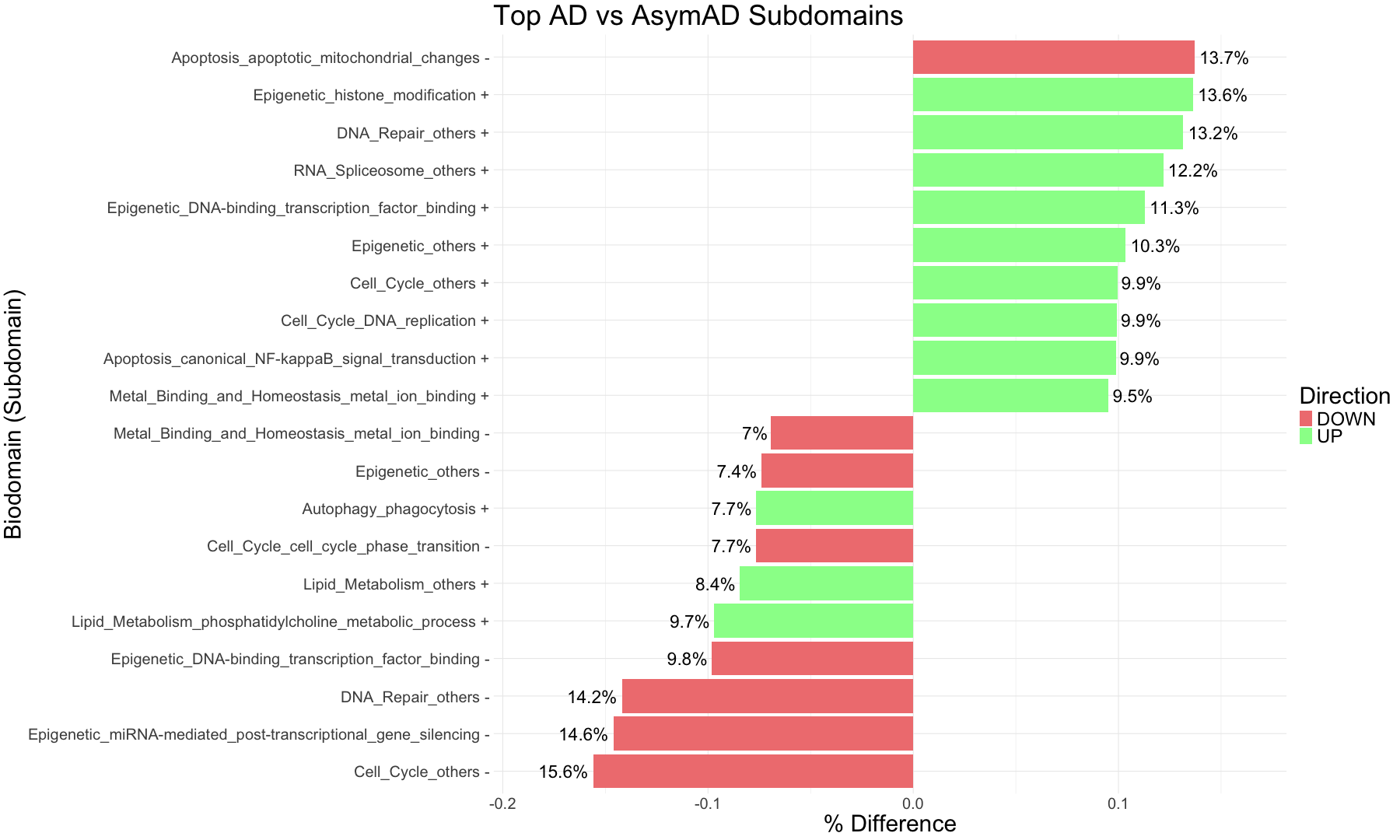
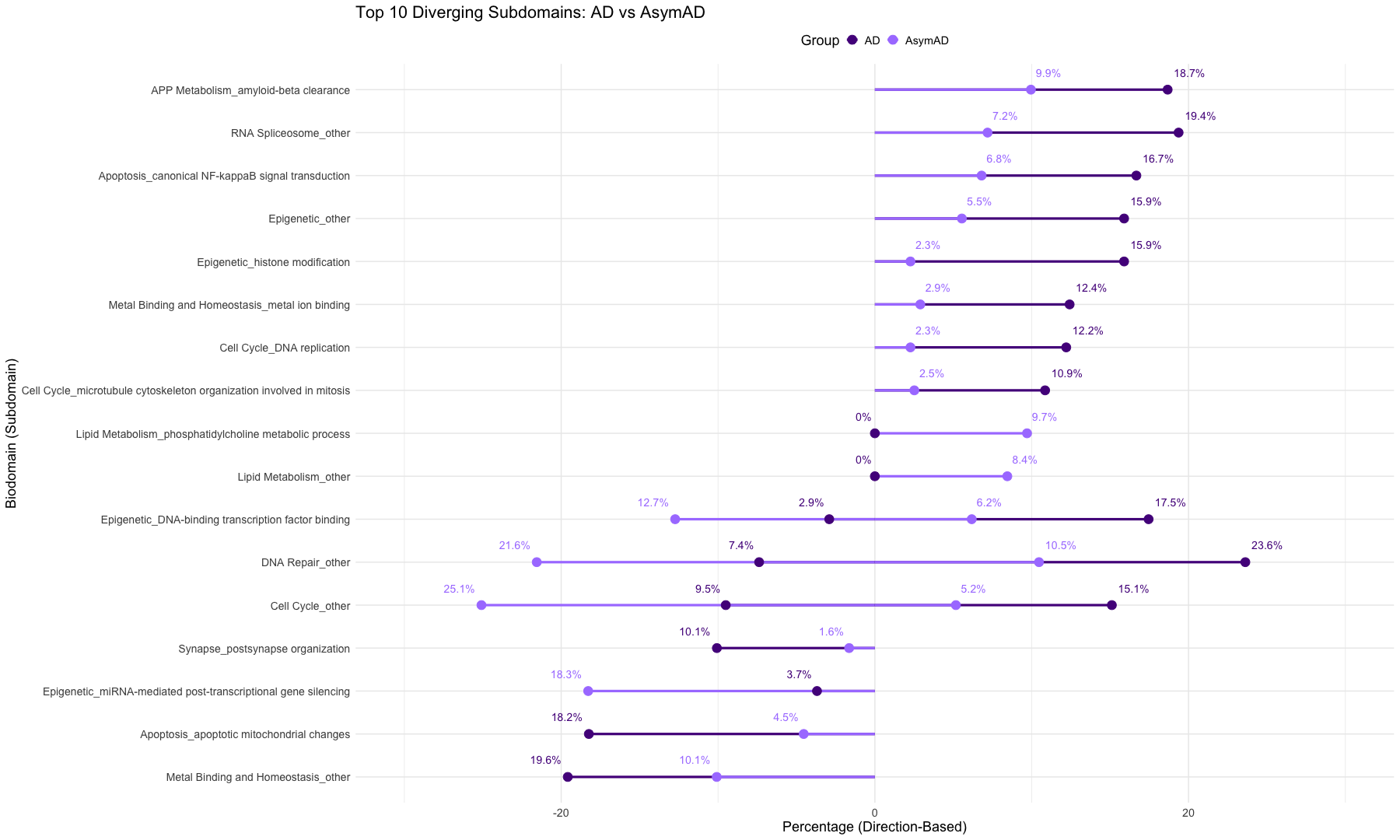
subset_data_subdomain_UP <- data_subdomain_new_UP %>% filter(UP_ratio_AD_female >= 0.10 | UP_ratio_AD_male >= 0.10 | UP_ratio_AsymAD_female >= 0.10 | UP_ratio_AsymAD_male >= 0.10)
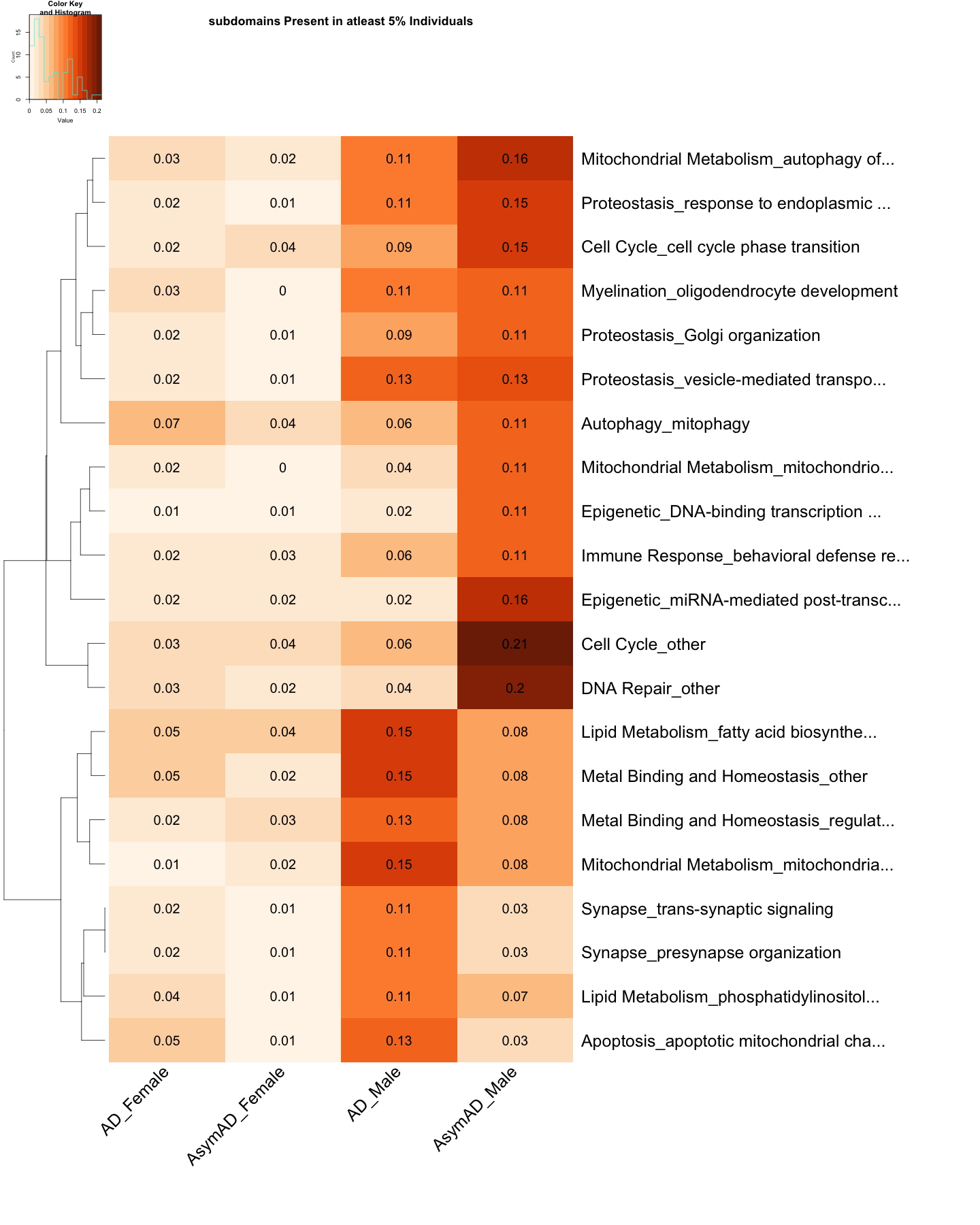
subset_data_subdomain_DOWN <- data_subdomain_new_DOWN %>% filter(DOWN_ratio_AD_female >= 0.10 | DOWN_ratio_AD_male >= 0.10 | DOWN_ratio_AsymAD_female >= 0.10 | DOWN_ratio_AsymAD_male >= 0.10)
rownames(subset_data_subdomain_UP) <- subset_data_subdomain_UP$Biodomain_Subdomain
rownames(subset_data_subdomain_DOWN) <- subset_data_subdomain_DOWN$Biodomain_Subdomain
subset_data_subdomain_UP <- subset_data_subdomain_UP[, -1]
subset_data_subdomain_DOWN <- subset_data_subdomain_DOWN[, -1]
# Replace NA values with 0 (or any other specific value)
subset_data_subdomain_UP[is.na(subset_data_subdomain_UP)] <- 0
subset_data_subdomain_DOWN[is.na(subset_data_subdomain_DOWN)] <- 0
head(subset_data_subdomain_UP, 3)
## UP_ratio_AD_female UP_ratio_AD_male
## APP Metabolism_amyloid-beta clearance 0.1015625 0.08510638
## APP Metabolism_amyloid-beta formation 0.1875000 0.00000000
## APP Metabolism_amyloid precursor prot... 0.0000000 0.10638298
## UP_ratio_AsymAD_female
## APP Metabolism_amyloid-beta clearance 0.050314465
## APP Metabolism_amyloid-beta formation 0.157232704
## APP Metabolism_amyloid precursor prot... 0.006289308
## UP_ratio_AsymAD_male
## APP Metabolism_amyloid-beta clearance 0.04918033
## APP Metabolism_amyloid-beta formation 0.03278689
## APP Metabolism_amyloid precursor prot... 0.08196721
head(subset_data_subdomain_DOWN, 3)
## DOWN_ratio_AD_female
## Apoptosis_apoptotic mitochondrial cha... 0.0546875
## Autophagy_mitophagy 0.0703125
## Cell Cycle_cell cycle phase transition 0.0234375
## DOWN_ratio_AD_male
## Apoptosis_apoptotic mitochondrial cha... 0.12765957
## Autophagy_mitophagy 0.06382979
## Cell Cycle_cell cycle phase transition 0.08510638
## DOWN_ratio_AsymAD_female
## Apoptosis_apoptotic mitochondrial cha... 0.01257862
## Autophagy_mitophagy 0.03773585
## Cell Cycle_cell cycle phase transition 0.03773585
## DOWN_ratio_AsymAD_male
## Apoptosis_apoptotic mitochondrial cha... 0.03278689
## Autophagy_mitophagy 0.11475410
## Cell Cycle_cell cycle phase transition 0.14754098
# Reorder the columns
subset_data_subdomain_UP <- subset_data_subdomain_UP[, c(
"UP_ratio_AD_female",
"UP_ratio_AsymAD_female",
"UP_ratio_AD_male",
"UP_ratio_AsymAD_male"
)]
subset_data_subdomain_DOWN <- subset_data_subdomain_DOWN[, c(
"DOWN_ratio_AD_female",
"DOWN_ratio_AsymAD_female",
"DOWN_ratio_AD_male",
"DOWN_ratio_AsymAD_male"
)]
# Assign custom column names
colnames(subset_data_subdomain_UP) <- c("AD_Female", "AsymAD_Female", "AD_Male", "AsymAD_Male")
colnames(subset_data_subdomain_DOWN) <- c("AD_Female", "AsymAD_Female", "AD_Male", "AsymAD_Male")
# Create the heatmap with filtered row names
UP <- heatmap.2(
as.matrix(subset_data_subdomain_UP),
Rowv = TRUE,
Colv = FALSE,
col = color_scale3,
scale = "none",
trace = "none",
margins = c(20, 50),
key = TRUE,
keysize = .50,
cexCol = 2.5,
srtCol = 45,
cexRow = 2.5,
cellnote=round(subset_data_subdomain_UP, 2),
notecex=2,
notecol="black",
dendrogram = "row",
main = "subdomains Present in atleast 5% Individuals"
)
# Create the heatmap with filtered row names
DOWN <- heatmap.2(
as.matrix(subset_data_subdomain_DOWN),
Rowv = TRUE,
Colv = FALSE,
col = color_scale6,
scale = "none",
trace = "none",
margins = c(20, 50),
key = TRUE,
keysize = .50,
cexCol = 2.5,
srtCol = 45,
cexRow = 2.5,
cellnote=round(subset_data_subdomain_DOWN, 2),
notecex=2,
notecol="black",
dendrogram = "row",
main = "subdomains Present in atleast 5% Individuals"
)