library(jpeg) # Load the .jpeg image img <- readJPEG("/Users/poddea/Desktop/ROSMAP_data_100623/Pathway_specific_Zscore.jpg") # Display the image plot(1:2, type = "n", xlab = "", ylab = "", axes = FALSE) rasterImage(img, 1, 1, 2, 2)
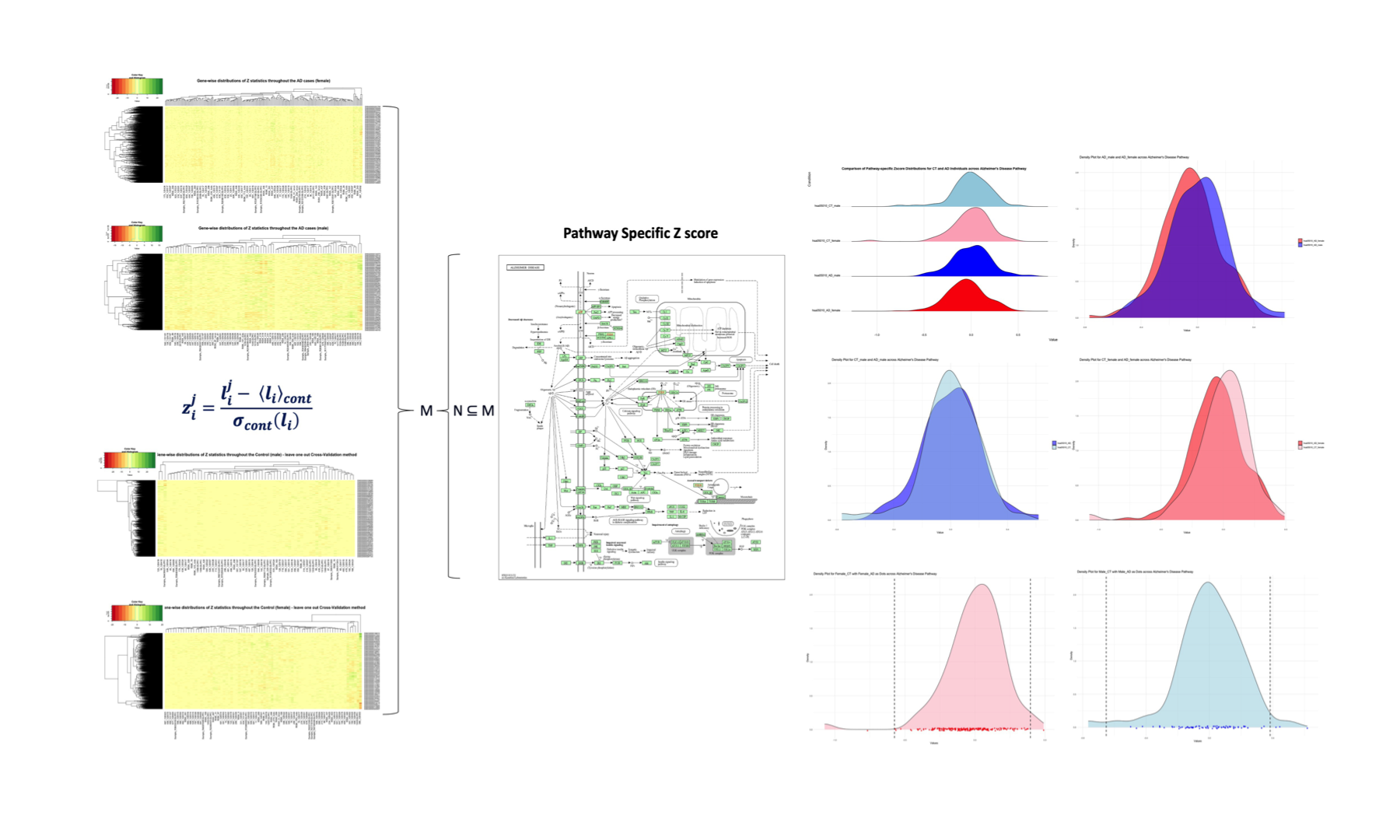
library(jpeg) # Load the .jpeg image img <- readJPEG("/Users/poddea/Desktop/ROSMAP_data_100623/Pathway_specific_Zscore.jpg") # Display the image plot(1:2, type = "n", xlab = "", ylab = "", axes = FALSE) rasterImage(img, 1, 1, 2, 2)
Retrieve human KEGG pathways and genes associated with each pathway:
#Create Individual Pathway Files:
#Open Protein-wise Zscore for CT, AD and AsymAD Individuals (Male and Female Separately):
library(dplyr)
library(tidyr) ## Open individual Zscore distribution files for CT and AD individuals zscore_CT_male <- read.delim("/Users/poddea/Desktop/ROSMAP_data_100623/TMT_proteomics/Round2/z_stat_result_CT_male.txt", header = TRUE, sep = "\t"); # Ensure column names to` match `IDs` in `zscore_` files zscore_CT_male <- zscore_CT_male %>% rename_with(~ gsub("X", "", .), starts_with("X")) dim(zscore_CT_male)
## [1] 7800 47
zscore_CT_female <- read.delim("/Users/poddea/Desktop/ROSMAP_data_100623/TMT_proteomics/Round2/z_stat_result_CT_female.txt", header = TRUE, sep = "\t"); # Ensure column names to` match `IDs` in `zscore_` files zscore_CT_female <- zscore_CT_female %>% rename_with(~ gsub("X", "", .), starts_with("X")) dim(zscore_CT_female)
## [1] 7800 69
zscore_AD_male <- read.delim("/Users/poddea/Desktop/ROSMAP_data_100623/TMT_proteomics/Round2/z_stat_result_AD_male.txt", header = TRUE, sep = "\t"); # Ensure column names to` match `IDs` in `zscore_` files zscore_AD_male <- zscore_AD_male %>% rename_with(~ gsub("X", "", .), starts_with("X")) dim(zscore_AD_male)
## [1] 7800 48
zscore_AD_female <- read.delim("/Users/poddea/Desktop/ROSMAP_data_100623/TMT_proteomics/Round2/z_stat_result_AD_female.txt", header = TRUE, sep = "\t"); # Ensure column names to` match `IDs` in `zscore_` files zscore_AD_female <- zscore_AD_female %>% rename_with(~ gsub("X", "", .), starts_with("X")) dim(zscore_AD_female)
## [1] 7800 128
zscore_AsymAD_male <- read.delim("/Users/poddea/Desktop/ROSMAP_data_100623/TMT_proteomics/Round2/z_stat_result_AsymAD_male.txt", header = TRUE, sep = "\t"); # Ensure column names to` match `IDs` in `zscore_` files zscore_AsymAD_male <- zscore_AsymAD_male %>% rename_with(~ gsub("X", "", .), starts_with("X")) dim(zscore_AsymAD_male)
## [1] 7800 62
zscore_AsymAD_female <- read.delim("/Users/poddea/Desktop/ROSMAP_data_100623/TMT_proteomics/Round2/z_stat_result_AsymAD_female.txt", header = TRUE, sep = "\t"); # Ensure column names to` match `IDs` in `zscore_` files zscore_AsymAD_female <- zscore_AsymAD_female %>% rename_with(~ gsub("X", "", .), starts_with("X")) dim(zscore_AsymAD_female)
## [1] 7800 160
Compare Gene-wise Zscore for CT and AD Individuals across Pathways (Example for Glycerophospholipid metabolism pathway):
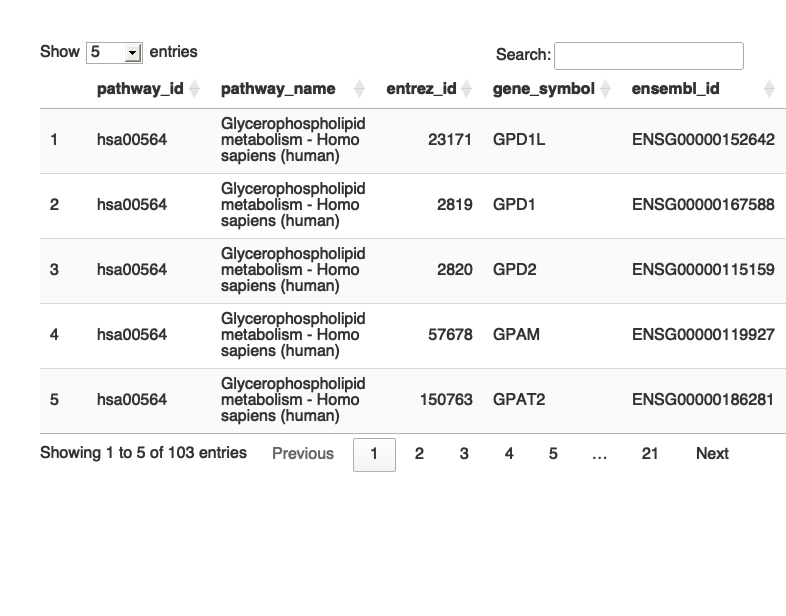
library(dplyr) library(tidyr) library(DT) setwd("/Users/poddea/Desktop/ROSMAP_data_100623/TMT_proteomics/Round2/Pathway_sprcific_analysis/kegg_hsa_all") # Open individual KEGG pathway file hsa00564 <- read.delim("hsa00564.txt", header = TRUE, sep = "\t"); dim(hsa00564)
## [1] 103 5
#head(hsa00564, 5) # Print the table using DT datatable(hsa00564, options = list(pageLength = 5, autoWidth = TRUE))
# Merge the data frames hsa00564_zscore_AD_female <- merge(hsa00564, zscore_AD_female, by.x = "gene_symbol", by.y = "Protein_name", all.x = TRUE) #head(hsa00564_zscore_AD_female, 2) # Print the table using DT datatable(hsa00564_zscore_AD_female, options = list(pageLength = 5, autoWidth = TRUE))

#write.table(hsa04710_zscore_AD_female,"/Users/poddea/Desktop/hsa04710_zscore_AD_female.txt",sep="\t",quote=F, row.name= FALSE) # Identify columns ending with "_Zscore" zscore_columns <- grep("_Zscore$", names(hsa00564_zscore_AD_female), value = TRUE) # Calculate the average for each of these columns average_zscores <- colMeans(hsa00564_zscore_AD_female[, zscore_columns], na.rm = TRUE) # Remove "_Zscore" suffix from the Sample_ID names sample_ids <- sub("_Zscore$", "", zscore_columns) # Create a new data frame with "Sample_ID" and the calculated averages Pathway_zscore_AD_female <- data.frame(Sample_ID = sample_ids, hsa00564_zscore_AD_female = average_zscores) #head(Pathway_zscore_AD_female) # Print the table using DT datatable(Pathway_zscore_AD_female, options = list(pageLength = 5, autoWidth = TRUE))
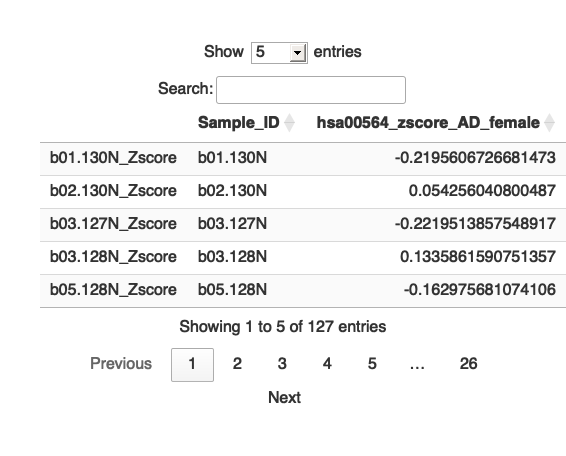
# Write the new data frame to a file #write.table(Pathway_zscore_AD_female, "/Users/poddea/Desktop/Pathway_zscore_AD_female.txt", sep = "\t", quote = FALSE, row.names = FALSE)
Compare Gene-wise Zscore for CT, AD and AsymAD Individuals across Pathways:
library(ggplot2)
## Warning: package 'ggplot2' was built under R version 4.5.2
library(gplots)
library(RColorBrewer) library(dplyr) library(tidyr) library(DT) ############################################################################################################################################################################ ## AD_female # Function to process each file process_kegg_file <- function(file_path, zscore_data) { # Read the KEGG pathway file kegg_data <- read.delim(file_path, header = TRUE, sep = "\t") # Merge with zscore_data merged_data <- merge(kegg_data, zscore_data, by.x = "gene_symbol", by.y = "Protein_name", all.x = TRUE) # Identify columns ending with "_Zscore" zscore_columns <- grep("_Zscore$", names(merged_data), value = TRUE) # Calculate Stouffer's combined Z-scores for each column stouffer_zscores <- apply(merged_data[, zscore_columns], 2, function(z_scores) { # Remove NA values z_scores <- na.omit(z_scores) # Calculate the Stouffer's Z-score stouffer_z <- sum(z_scores) / sqrt(length(z_scores)) return(stouffer_z) }) # Remove "_Zscore" suffix from the Sample_ID names sample_ids <- sub("_Zscore$", "", zscore_columns) # Create a new data frame with "Sample_ID" and the calculated Stouffer's Z-scores pathway_id <- unique(kegg_data$pathway_id)[1] colname <- paste0(pathway_id, "_Zscore") results_df <- data.frame(Sample_ID = sample_ids) results_df[[colname]] <- stouffer_zscores return(results_df) } # Path to the directory containing hsa*.txt files dir_path <- "/Users/poddea/Desktop/ROSMAP_data_100623/TMT_proteomics/Round2/Pathway_sprcific_analysis/kegg_hsa_all" # Read the zscore_AD_female data #zscore_AD_female <- read.delim("zscore_AD_female.txt", header = TRUE, sep = "\t") # List all hsa*.txt files in the directory kegg_files <- list.files(path = dir_path, pattern = "^hsa.*\\.txt$", full.names = TRUE) # Initialize an empty list to store the results all_results <- list() # Process each KEGG file for (file in kegg_files) { result <- process_kegg_file(file, zscore_AD_female) all_results[[file]] <- result } # Combine all results into one data frame by merging them on Sample_ID final_results_AD_female <- Reduce(function(x, y) merge(x, y, by = "Sample_ID", all = TRUE), all_results) # Write the final results to a file write.table(final_results_AD_female, "Pathway_zscore_AD_female_summary.txt", sep = "\t", quote = FALSE, row.names = FALSE) ########################################################################################################################################################### # Transpose the file to count 1st and 99th percentile of the Z score distributions in individual pathway final_results_AD_female_t <- as.data.frame(t(final_results_AD_female[,-1])) colnames(final_results_AD_female_t) <- final_results_AD_female$Sample_ID # Convert the row names to a new column named "Pathway_ID" final_results_AD_female_t$Pathway_ID <- rownames(final_results_AD_female_t) # Reorder the columns so that "Pathway_ID" comes first final_results_AD_female_t <- final_results_AD_female_t[, c("Pathway_ID", names(final_results_AD_female_t)[-ncol(final_results_AD_female_t)])] # Reset the row names as they are no longer needed rownames(final_results_AD_female_t) <- NULL # Print the table using DT datatable(final_results_AD_female_t, options = list(pageLength = 5, autoWidth = TRUE))

########################################################################################################################################################### # Plot tha data # Set rownames to Sample_ID and remove Sample_ID column rownames(final_results_AD_female) <- final_results_AD_female$Sample_ID final_results_AD_female <- final_results_AD_female[, -1] # Remove columns with NA values final_results_AD_female <- final_results_AD_female[, colSums(is.na(final_results_AD_female)) == 0] # Set up the color scale using RColorBrewer color_scale <- colorRampPalette(brewer.pal(9, "RdYlGn")) # Plot the heatmap using heatmap.2 heatmap.2( as.matrix(final_results_AD_female), Rowv = TRUE, Colv = TRUE, col = color_scale, scale = "column", # Scale rows (genes) to Z-scores trace = "none", # Turn off row and column annotations margins = c(20, 20), # Set margins for row and column labels #col = cm.colors(256), # Set the color map (256 colors) key = TRUE, # Include a color key keysize = 1, # Size of the color key cexCol = 1, # Set column label size cexRow = 0.8, # Set row label size dendrogram = "both", # Show both row and column dendrograms main="Pathway-specific Z score for AD Individuals (female)" )

############################################################################################################################################################################ ## Count the number of gene identified in each pathway # Function to process each file process_kegg_file <- function(file_path, zscore_data) { # Read the KEGG pathway file kegg_data <- read.delim(file_path, header = TRUE, sep = "\t") # Merge with zscore_data merged_data <- merge(kegg_data, zscore_data, by.x = "gene_symbol", by.y = "Protein_name", all.x = TRUE) # Identify columns ending with "_Zscore" zscore_columns <- grep("_Zscore$", names(merged_data), value = TRUE) # Calculate the number of non-NA values (mapped genes) for each sample gene_counts <- apply(merged_data[, zscore_columns], 2, function(z_scores) { return(sum(!is.na(z_scores))) }) # Remove "_Zscore" suffix from the Sample_ID names sample_ids <- sub("_Zscore$", "", zscore_columns) # Create a new data frame with "Sample_ID" and the number of mapped genes pathway_id <- unique(kegg_data$pathway_id)[1] colname <- paste0(pathway_id, "_gene_count") gene_count_df <- data.frame(Sample_ID = sample_ids) gene_count_df[[colname]] <- gene_counts return(gene_count_df) } # Path to the directory containing hsa*.txt files dir_path <- "/Users/poddea/Desktop/ROSMAP_data_100623/TMT_proteomics/Round2/Pathway_sprcific_analysis/kegg_hsa_all" # Read the zscore_AD_female data # zscore_AD_female <- read.delim("zscore_AD_female.txt", header = TRUE, sep = "\t") # List all hsa*.txt files in the directory kegg_files <- list.files(path = dir_path, pattern = "^hsa.*\\.txt$", full.names = TRUE) # Initialize an empty list to store the results all_gene_counts <- list() # Process each KEGG file to count the number of mapped genes for (file in kegg_files) { gene_count_result <- process_kegg_file(file, zscore_AD_female) all_gene_counts[[file]] <- gene_count_result } # Combine all gene count results into one data frame by merging them on Sample_ID final_gene_counts_AD_female <- Reduce(function(x, y) merge(x, y, by = "Sample_ID", all = TRUE), all_gene_counts) # Write the final gene counts to a file write.table(final_gene_counts_AD_female, "Pathway_gene_count_AD_female_summary.txt", sep = "\t", quote = FALSE, row.names = FALSE) ############################################################################################################################################################################ ############################################################################################################################################################################ ## AD_male # Function to process each file process_kegg_file <- function(file_path, zscore_data) { # Read the KEGG pathway file kegg_data <- read.delim(file_path, header = TRUE, sep = "\t") # Merge with zscore_data merged_data <- merge(kegg_data, zscore_data, by.x = "gene_symbol", by.y = "Protein_name", all.x = TRUE) # Identify columns ending with "_Zscore" zscore_columns <- grep("_Zscore$", names(merged_data), value = TRUE) # Calculate Stouffer's combined Z-scores for each column stouffer_zscores <- apply(merged_data[, zscore_columns], 2, function(z_scores) { # Remove NA values z_scores <- na.omit(z_scores) # Calculate the Stouffer's Z-score stouffer_z <- sum(z_scores) / sqrt(length(z_scores)) return(stouffer_z) }) # Remove "_Zscore" suffix from the Sample_ID names sample_ids <- sub("_Zscore$", "", zscore_columns) # Create a new data frame with "Sample_ID" and the calculated Stouffer's Z-scores pathway_id <- unique(kegg_data$pathway_id)[1] colname <- paste0(pathway_id, "_Zscore") results_df <- data.frame(Sample_ID = sample_ids) results_df[[colname]] <- stouffer_zscores return(results_df) } # Path to the directory containing hsa*.txt files dir_path <- "/Users/poddea/Desktop/ROSMAP_data_100623/TMT_proteomics/Round2/Pathway_sprcific_analysis/kegg_hsa_all" # Read the zscore_AD_female data #zscore_AD_female <- read.delim("zscore_AD_male.txt", header = TRUE, sep = "\t") # List all hsa*.txt files in the directory kegg_files <- list.files(path = dir_path, pattern = "^hsa.*\\.txt$", full.names = TRUE) # Initialize an empty list to store the results all_results <- list() # Process each KEGG file for (file in kegg_files) { result <- process_kegg_file(file, zscore_AD_male) all_results[[file]] <- result } # Combine all results into one data frame by merging them on Sample_ID final_results_AD_male <- Reduce(function(x, y) merge(x, y, by = "Sample_ID", all = TRUE), all_results) # Write the final results to a file write.table(final_results_AD_male, "Pathway_zscore_AD_male_summary.txt", sep = "\t", quote = FALSE, row.names = FALSE) ########################################################################################################################################################### # Transpose the file to count 1st and 99th percentile of the Z score distributions in individual pathway final_results_AD_male_t <- as.data.frame(t(final_results_AD_male[,-1])) colnames(final_results_AD_male_t) <- final_results_AD_male$Sample_ID # Convert the row names to a new column named "Pathway_ID" final_results_AD_male_t$Pathway_ID <- rownames(final_results_AD_male_t) # Reorder the columns so that "Pathway_ID" comes first final_results_AD_male_t <- final_results_AD_male_t[, c("Pathway_ID", names(final_results_AD_male_t)[-ncol(final_results_AD_male_t)])] # Reset the row names as they are no longer needed rownames(final_results_AD_male_t) <- NULL # Print the table using DT datatable(final_results_AD_male_t, options = list(pageLength = 5, autoWidth = TRUE))
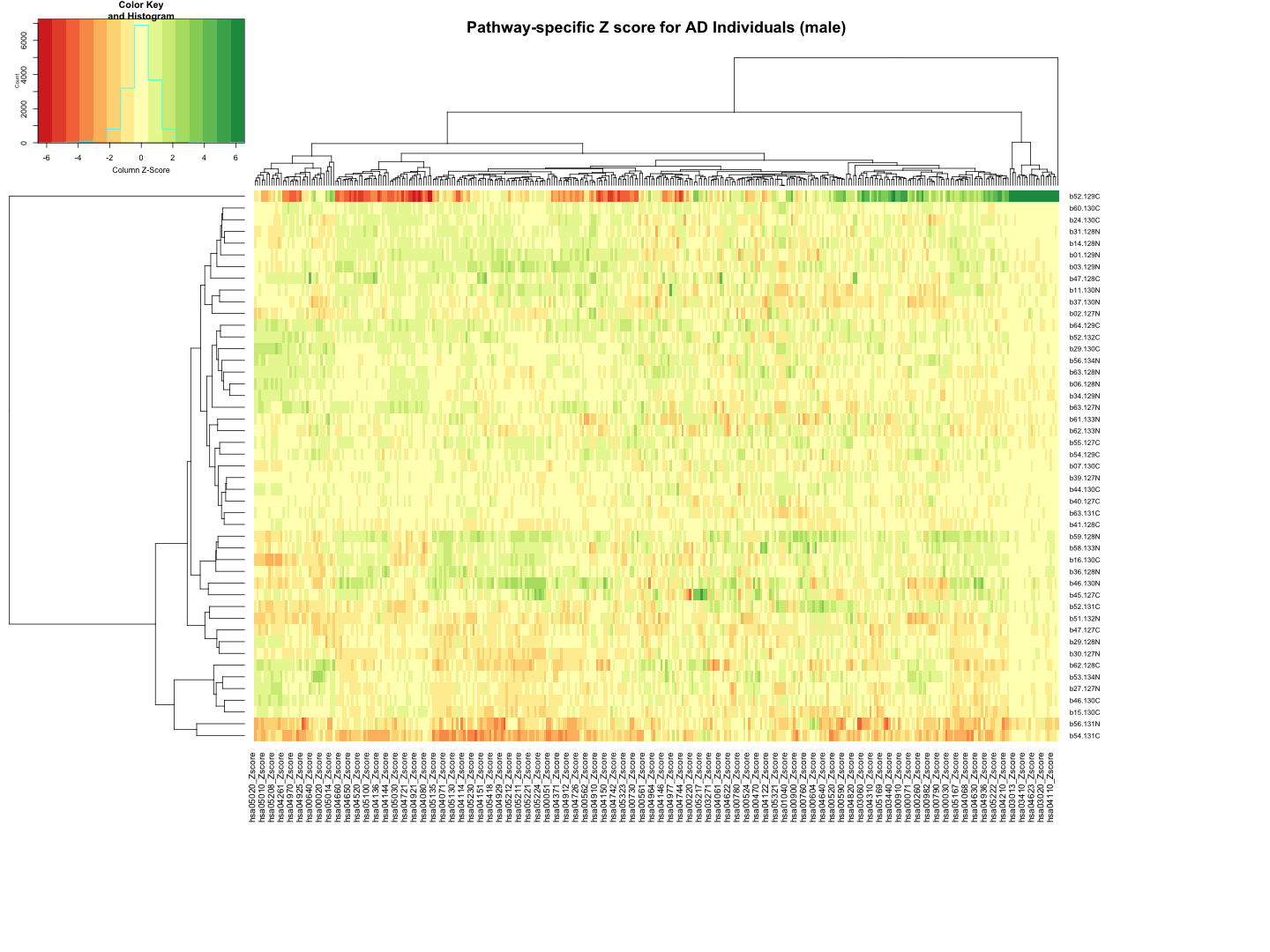
########################################################################################################################################################### # Plot tha data # Set rownames to Sample_ID and remove Sample_ID column rownames(final_results_AD_male) <- final_results_AD_male$Sample_ID final_results_AD_male <- final_results_AD_male[, -1] # Remove columns with NA values final_results_AD_male <- final_results_AD_male[, colSums(is.na(final_results_AD_male)) == 0] # Set up the color scale using RColorBrewer color_scale <- colorRampPalette(brewer.pal(9, "RdYlGn")) # Plot the heatmap using heatmap.2 heatmap.2( as.matrix(final_results_AD_male), Rowv = TRUE, Colv = TRUE, col = color_scale, scale = "column", # Scale rows (genes) to Z-scores trace = "none", # Turn off row and column annotations margins = c(20, 20), # Set margins for row and column labels #col = cm.colors(256), # Set the color map (256 colors) key = TRUE, # Include a color key keysize = 1, # Size of the color key cexCol = 1, # Set column label size cexRow = 0.8, # Set row label size dendrogram = "both", # Show both row and column dendrograms main="Pathway-specific Z score for AD Individuals (male)" )
############################################################################################################################################################################ ############################################################################################################################################################################ ## AsymAD_female # Function to process each file process_kegg_file <- function(file_path, zscore_data) { # Read the KEGG pathway file kegg_data <- read.delim(file_path, header = TRUE, sep = "\t") # Merge with zscore_data merged_data <- merge(kegg_data, zscore_data, by.x = "gene_symbol", by.y = "Protein_name", all.x = TRUE) # Identify columns ending with "_Zscore" zscore_columns <- grep("_Zscore$", names(merged_data), value = TRUE) # Calculate Stouffer's combined Z-scores for each column stouffer_zscores <- apply(merged_data[, zscore_columns], 2, function(z_scores) { # Remove NA values z_scores <- na.omit(z_scores) # Calculate the Stouffer's Z-score stouffer_z <- sum(z_scores) / sqrt(length(z_scores)) return(stouffer_z) }) # Remove "_Zscore" suffix from the Sample_ID names sample_ids <- sub("_Zscore$", "", zscore_columns) # Create a new data frame with "Sample_ID" and the calculated Stouffer's Z-scores pathway_id <- unique(kegg_data$pathway_id)[1] colname <- paste0(pathway_id, "_Zscore") results_df <- data.frame(Sample_ID = sample_ids) results_df[[colname]] <- stouffer_zscores return(results_df) } # Path to the directory containing hsa*.txt files dir_path <- "/Users/poddea/Desktop/ROSMAP_data_100623/TMT_proteomics/Round2/Pathway_sprcific_analysis/kegg_hsa_all" # Read the zscore_AsymAD_female data #zscore_AsymAD_female <- read.delim("zscore_AsymAD_female.txt", header = TRUE, sep = "\t") # List all hsa*.txt files in the directory kegg_files <- list.files(path = dir_path, pattern = "^hsa.*\\.txt$", full.names = TRUE) # Initialize an empty list to store the results all_results <- list() # Process each KEGG file for (file in kegg_files) { result <- process_kegg_file(file, zscore_AsymAD_female) all_results[[file]] <- result } # Combine all results into one data frame by merging them on Sample_ID final_results_AsymAD_female <- Reduce(function(x, y) merge(x, y, by = "Sample_ID", all = TRUE), all_results) # Write the final results to a file write.table(final_results_AsymAD_female, "Pathway_zscore_AsymAD_female_summary.txt", sep = "\t", quote = FALSE, row.names = FALSE) ########################################################################################################################################################### # Transpose the file to count 1st and 99th percentile of the Z score distributions in individual pathway final_results_AsymAD_female_t <- as.data.frame(t(final_results_AsymAD_female[,-1])) colnames(final_results_AsymAD_female_t) <- final_results_AsymAD_female$Sample_ID # Convert the row names to a new column named "Pathway_ID" final_results_AsymAD_female_t$Pathway_ID <- rownames(final_results_AsymAD_female_t) # Reorder the columns so that "Pathway_ID" comes first final_results_AsymAD_female_t <- final_results_AsymAD_female_t[, c("Pathway_ID", names(final_results_AsymAD_female_t)[-ncol(final_results_AsymAD_female_t)])] # Reset the row names as they are no longer needed rownames(final_results_AsymAD_female_t) <- NULL # Print the table using DT datatable(final_results_AsymAD_female_t, options = list(pageLength = 5, autoWidth = TRUE))

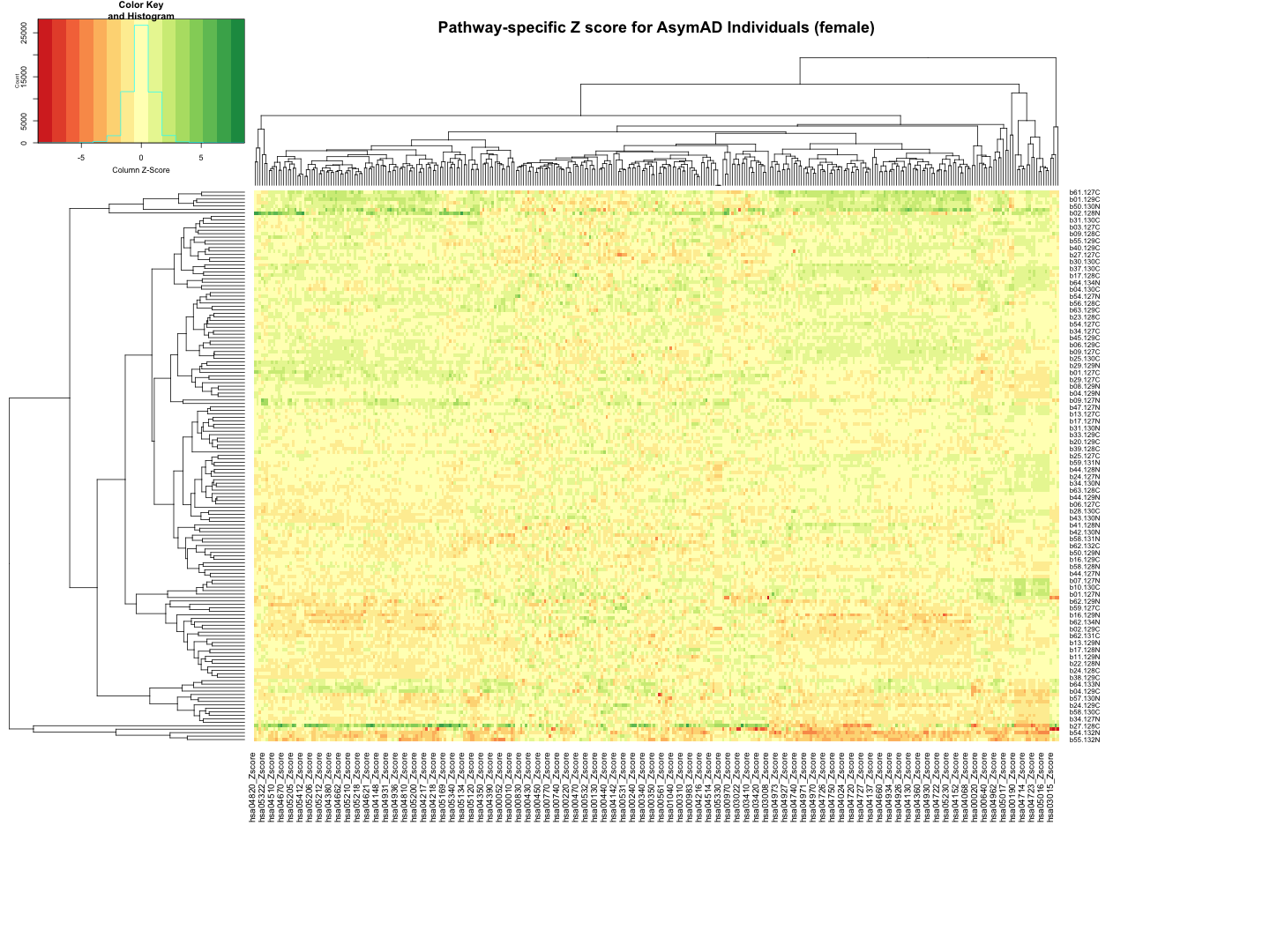
########################################################################################################################################################### # Plot tha data # Set rownames to Sample_ID and remove Sample_ID column rownames(final_results_AsymAD_female) <- final_results_AsymAD_female$Sample_ID final_results_AsymAD_female <- final_results_AsymAD_female[, -1] # Remove columns with NA values final_results_AsymAD_female <- final_results_AsymAD_female[, colSums(is.na(final_results_AsymAD_female)) == 0] # Set up the color scale using RColorBrewer color_scale <- colorRampPalette(brewer.pal(9, "RdYlGn")) # Plot the heatmap using heatmap.2 heatmap.2( as.matrix(final_results_AsymAD_female), Rowv = TRUE, Colv = TRUE, col = color_scale, scale = "column", # Scale rows (genes) to Z-scores trace = "none", # Turn off row and column annotations margins = c(20, 20), # Set margins for row and column labels #col = cm.colors(256), # Set the color map (256 colors) key = TRUE, # Include a color key keysize = 1, # Size of the color key cexCol = 1, # Set column label size cexRow = 0.8, # Set row label size dendrogram = "both", # Show both row and column dendrograms main="Pathway-specific Z score for AsymAD Individuals (female)" )
############################################################################################################################################################################ ############################################################################################################################################################################ ## AsymAD_male # Function to process each file process_kegg_file <- function(file_path, zscore_data) { # Read the KEGG pathway file kegg_data <- read.delim(file_path, header = TRUE, sep = "\t") # Merge with zscore_data merged_data <- merge(kegg_data, zscore_data, by.x = "gene_symbol", by.y = "Protein_name", all.x = TRUE) # Identify columns ending with "_Zscore" zscore_columns <- grep("_Zscore$", names(merged_data), value = TRUE) # Calculate Stouffer's combined Z-scores for each column stouffer_zscores <- apply(merged_data[, zscore_columns], 2, function(z_scores) { # Remove NA values z_scores <- na.omit(z_scores) # Calculate the Stouffer's Z-score stouffer_z <- sum(z_scores) / sqrt(length(z_scores)) return(stouffer_z) }) # Remove "_Zscore" suffix from the Sample_ID names sample_ids <- sub("_Zscore$", "", zscore_columns) # Create a new data frame with "Sample_ID" and the calculated Stouffer's Z-scores pathway_id <- unique(kegg_data$pathway_id)[1] colname <- paste0(pathway_id, "_Zscore") results_df <- data.frame(Sample_ID = sample_ids) results_df[[colname]] <- stouffer_zscores return(results_df) } # Path to the directory containing hsa*.txt files dir_path <- "/Users/poddea/Desktop/ROSMAP_data_100623/TMT_proteomics/Round2/Pathway_sprcific_analysis/kegg_hsa_all" # Read the zscore_AsymAD_male data #zscore_AsymAD_male <- read.delim("zscore_AsymAD_male.txt", header = TRUE, sep = "\t") # List all hsa*.txt files in the directory kegg_files <- list.files(path = dir_path, pattern = "^hsa.*\\.txt$", full.names = TRUE) # Initialize an empty list to store the results all_results <- list() # Process each KEGG file for (file in kegg_files) { result <- process_kegg_file(file, zscore_AsymAD_male) all_results[[file]] <- result } # Combine all results into one data frame by merging them on Sample_ID final_results_AsymAD_male <- Reduce(function(x, y) merge(x, y, by = "Sample_ID", all = TRUE), all_results) # Write the final results to a file write.table(final_results_AsymAD_male, "Pathway_zscore_AsymAD_male_summary.txt", sep = "\t", quote = FALSE, row.names = FALSE) ########################################################################################################################################################### # Transpose the file to count 1st and 99th percentile of the Z score distributions in individual pathway final_results_AsymAD_male_t <- as.data.frame(t(final_results_AsymAD_male[,-1])) colnames(final_results_AsymAD_male_t) <- final_results_AsymAD_male$Sample_ID # Convert the row names to a new column named "Pathway_ID" final_results_AsymAD_male_t$Pathway_ID <- rownames(final_results_AsymAD_male_t) # Reorder the columns so that "Pathway_ID" comes first final_results_AsymAD_male_t <- final_results_AsymAD_male_t[, c("Pathway_ID", names(final_results_AsymAD_male_t)[-ncol(final_results_AsymAD_male_t)])] # Reset the row names as they are no longer needed rownames(final_results_AsymAD_male_t) <- NULL # Print the table using DT datatable(final_results_AsymAD_male_t, options = list(pageLength = 5, autoWidth = TRUE))

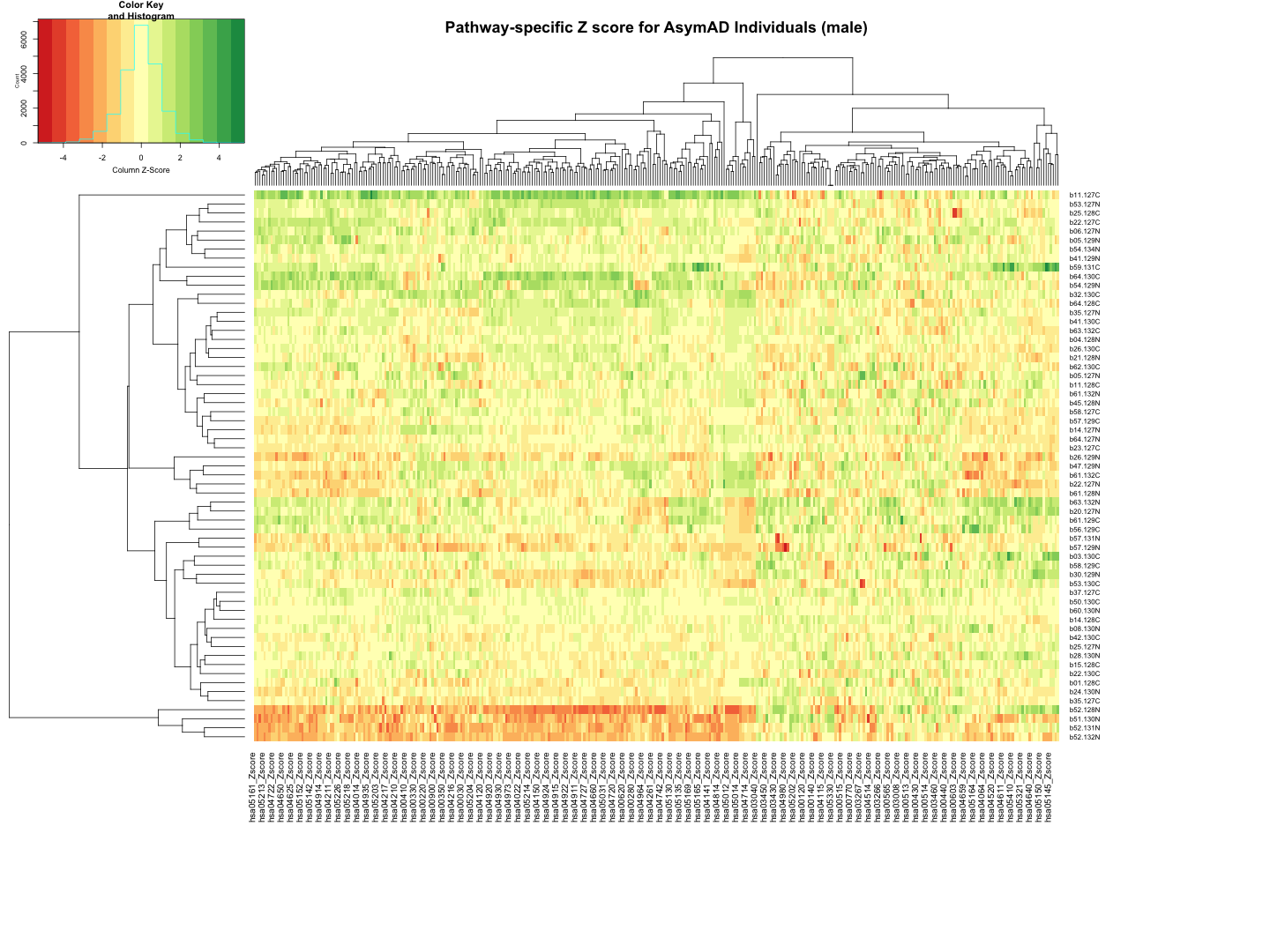
########################################################################################################################################################### # Plot tha data # Set rownames to Sample_ID and remove Sample_ID column rownames(final_results_AsymAD_male) <- final_results_AsymAD_male$Sample_ID final_results_AsymAD_male <- final_results_AsymAD_male[, -1] # Remove columns with NA values final_results_AsymAD_male <- final_results_AsymAD_male[, colSums(is.na(final_results_AsymAD_male)) == 0] # Set up the color scale using RColorBrewer color_scale <- colorRampPalette(brewer.pal(9, "RdYlGn")) # Plot the heatmap using heatmap.2 heatmap.2( as.matrix(final_results_AsymAD_male), Rowv = TRUE, Colv = TRUE, col = color_scale, scale = "column", # Scale rows (genes) to Z-scores trace = "none", # Turn off row and column annotations margins = c(20, 20), # Set margins for row and column labels #col = cm.colors(256), # Set the color map (256 colors) key = TRUE, # Include a color key keysize = 1, # Size of the color key cexCol = 1, # Set column label size cexRow = 0.8, # Set row label size dendrogram = "both", # Show both row and column dendrograms main="Pathway-specific Z score for AsymAD Individuals (male)" )
############################################################################################################################################################################ ############################################################################################################################################################################ ## CT_female # Function to process each file process_kegg_file <- function(file_path, zscore_data) { # Read the KEGG pathway file kegg_data <- read.delim(file_path, header = TRUE, sep = "\t") # Merge with zscore_data merged_data <- merge(kegg_data, zscore_data, by.x = "gene_symbol", by.y = "Protein_name", all.x = TRUE) # Identify columns ending with "_Zscore" zscore_columns <- grep("_Zscore$", names(merged_data), value = TRUE) # Calculate Stouffer's combined Z-scores for each column stouffer_zscores <- apply(merged_data[, zscore_columns], 2, function(z_scores) { # Remove NA values z_scores <- na.omit(z_scores) # Calculate the Stouffer's Z-score stouffer_z <- sum(z_scores) / sqrt(length(z_scores)) return(stouffer_z) }) # Remove "_Zscore" suffix from the Sample_ID names sample_ids <- sub("_Zscore$", "", zscore_columns) # Create a new data frame with "Sample_ID" and the calculated Stouffer's Z-scores pathway_id <- unique(kegg_data$pathway_id)[1] colname <- paste0(pathway_id, "_Zscore") results_df <- data.frame(Sample_ID = sample_ids) results_df[[colname]] <- stouffer_zscores return(results_df) } # Path to the directory containing hsa*.txt files dir_path <- "/Users/poddea/Desktop/ROSMAP_data_100623/TMT_proteomics/Round2/Pathway_sprcific_analysis/kegg_hsa_all" # Read the zscore_AD_female data #zscore_AD_female <- read.delim("zscore_AD_female.txt", header = TRUE, sep = "\t") # List all hsa*.txt files in the directory kegg_files <- list.files(path = dir_path, pattern = "^hsa.*\\.txt$", full.names = TRUE) # Initialize an empty list to store the results all_results <- list() # Process each KEGG file for (file in kegg_files) { result <- process_kegg_file(file, zscore_CT_female) all_results[[file]] <- result } # Combine all results into one data frame by merging them on Sample_ID final_results_CT_female <- Reduce(function(x, y) merge(x, y, by = "Sample_ID", all = TRUE), all_results) # Write the final results to a file write.table(final_results_CT_female, "Pathway_zscore_CT_female_summary.txt", sep = "\t", quote = FALSE, row.names = FALSE) ########################################################################################################################################################### # Transpose the file to count 1st and 99th percentile of the Z score distributions in individual pathway final_results_CT_female_t <- as.data.frame(t(final_results_CT_female[,-1])) colnames(final_results_CT_female_t) <- final_results_CT_female$Sample_ID # Convert the row names to a new column named "Pathway_ID" final_results_CT_female_t$Pathway_ID <- rownames(final_results_CT_female_t) # Reorder the columns so that "Pathway_ID" comes first final_results_CT_female_t <- final_results_CT_female_t[, c("Pathway_ID", names(final_results_CT_female_t)[-ncol(final_results_CT_female_t)])] # Reset the row names as they are no longer needed rownames(final_results_CT_female_t) <- NULL # Print the table using DT datatable(final_results_CT_female_t, options = list(pageLength = 5, autoWidth = TRUE))

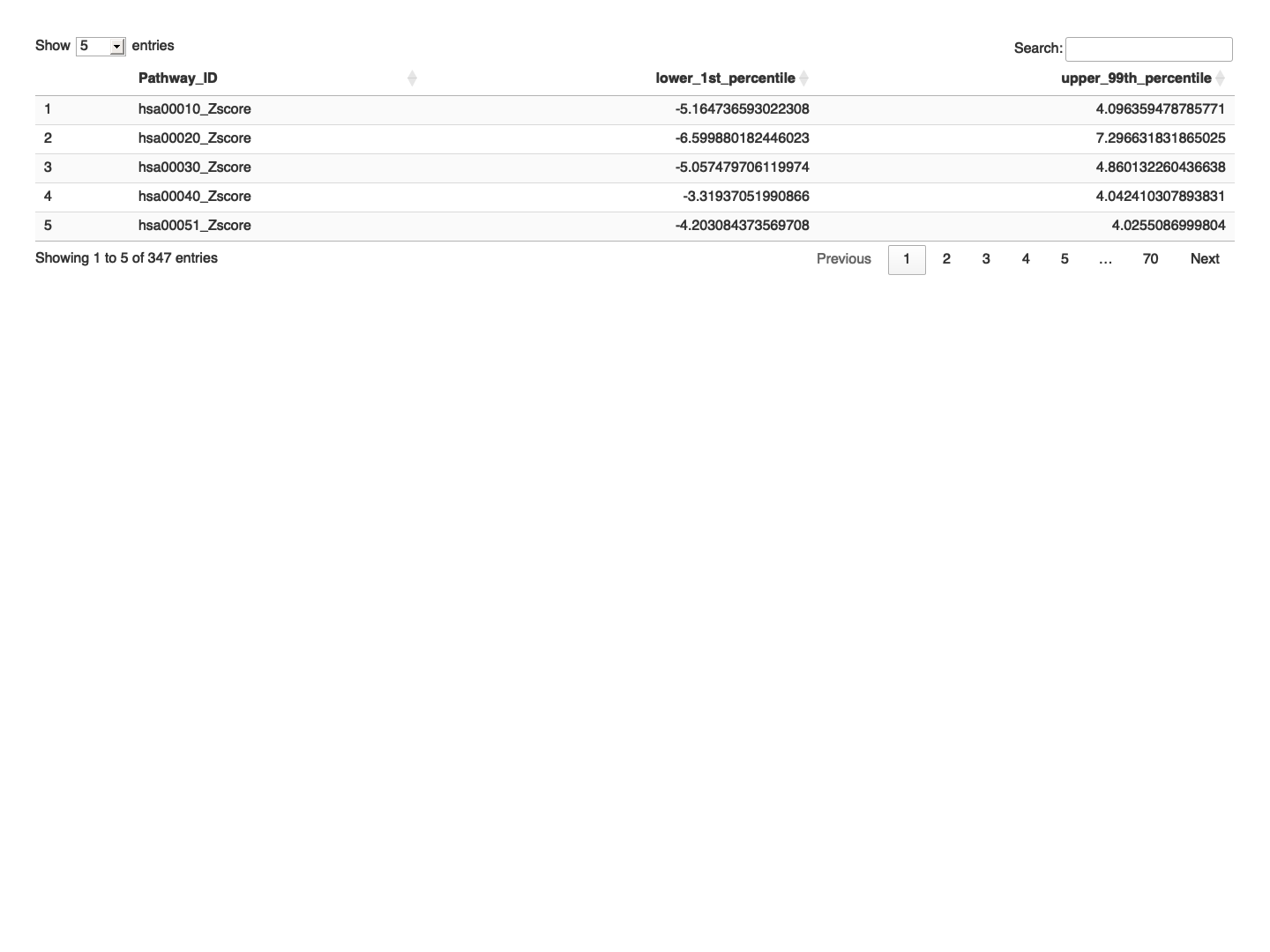
# Function to calculate percentiles calc_percentiles <- function(x) { lower <- quantile(x, probs = 0.01, na.rm = TRUE) upper <- quantile(x, probs = 0.99, na.rm = TRUE) return(c(lower, upper)) } # Apply the function to each row percentiles <- t(apply(final_results_CT_female_t[,-1], 1, calc_percentiles)) # Add the percentiles to the data frame final_results_CT_female_t_percentile <- cbind(final_results_CT_female_t, lower_1st_percentile = percentiles[, 1], upper_99th_percentile = percentiles[, 2]) # Define a dataframe with thresold values for each gene within the control sample zscored_threshold_female <- final_results_CT_female_t_percentile[,c("Pathway_ID", "lower_1st_percentile", "upper_99th_percentile")] # Write the z-scored and 5th and 95th Percentiles for Each Gene to a text file write.table(zscored_threshold_female, "zscored_percentiles_pathway_data_DLPFC_contorl_female.txt", sep="\t", quote=FALSE, row.names=FALSE) # Print the table using DT datatable(zscored_threshold_female, options = list(pageLength = 5, autoWidth = TRUE))
############################################################################################################################################################ # Plot the data # Set rownames to Sample_ID and remove Sample_ID column rownames(final_results_CT_female) <- final_results_CT_female$Sample_ID final_results_CT_female <- final_results_CT_female[, -1] # Remove columns with NA values final_results_CT_female <- final_results_CT_female[, colSums(is.na(final_results_CT_female)) == 0] # Set up the color scale using RColorBrewer color_scale <- colorRampPalette(brewer.pal(9, "RdYlGn")) # Plot the heatmap using heatmap.2 heatmap.2( as.matrix(final_results_CT_female), Rowv = TRUE, Colv = TRUE, col = color_scale, scale = "column", # Scale rows (genes) to Z-scores trace = "none", # Turn off row and column annotations margins = c(20, 20), # Set margins for row and column labels #col = cm.colors(256), # Set the color map (256 colors) key = TRUE, # Include a color key keysize = 1, # Size of the color key cexCol = 1, # Set column label size cexRow = 0.8, # Set row label size dendrogram = "both", # Show both row and column dendrograms main="Pathway-specific Z score for CT Individuals (female)" )
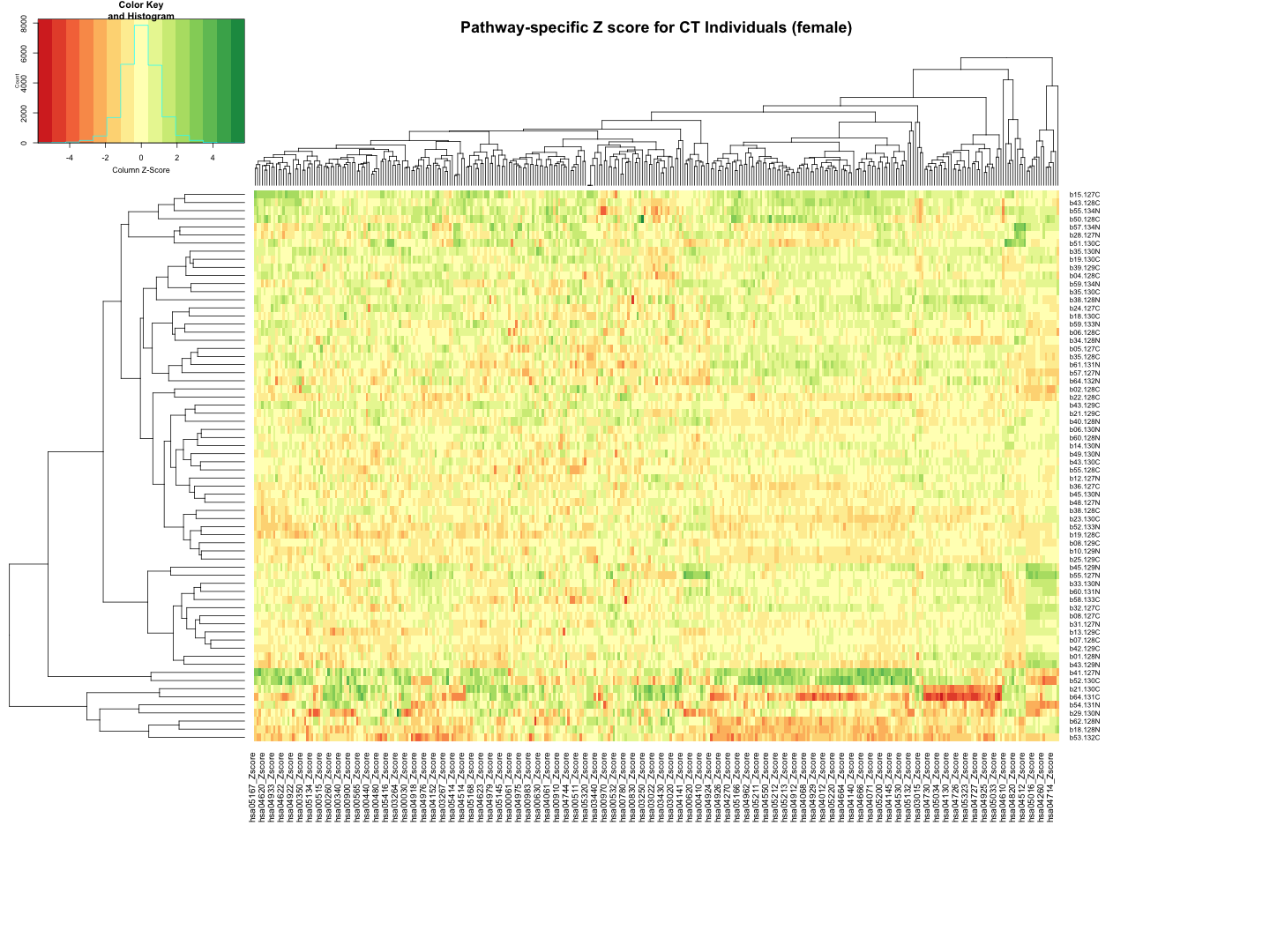
############################################################################################################################################################################ ############################################################################################################################################################################ ## CT_male # Function to process each file process_kegg_file <- function(file_path, zscore_data) { # Read the KEGG pathway file kegg_data <- read.delim(file_path, header = TRUE, sep = "\t") # Merge with zscore_data merged_data <- merge(kegg_data, zscore_data, by.x = "gene_symbol", by.y = "Protein_name", all.x = TRUE) # Identify columns ending with "_Zscore" zscore_columns <- grep("_Zscore$", names(merged_data), value = TRUE) # Calculate Stouffer's combined Z-scores for each column stouffer_zscores <- apply(merged_data[, zscore_columns], 2, function(z_scores) { # Remove NA values z_scores <- na.omit(z_scores) # Calculate the Stouffer's Z-score stouffer_z <- sum(z_scores) / sqrt(length(z_scores)) return(stouffer_z) }) # Remove "_Zscore" suffix from the Sample_ID names sample_ids <- sub("_Zscore$", "", zscore_columns) # Create a new data frame with "Sample_ID" and the calculated Stouffer's Z-scores pathway_id <- unique(kegg_data$pathway_id)[1] colname <- paste0(pathway_id, "_Zscore") results_df <- data.frame(Sample_ID = sample_ids) results_df[[colname]] <- stouffer_zscores return(results_df) } # Path to the directory containing hsa*.txt files dir_path <- "/Users/poddea/Desktop/ROSMAP_data_100623/TMT_proteomics/Round2/Pathway_sprcific_analysis/kegg_hsa_all" # Read the zscore_AD_female data #zscore_AD_female <- read.delim("zscore_AD_female.txt", header = TRUE, sep = "\t") # List all hsa*.txt files in the directory kegg_files <- list.files(path = dir_path, pattern = "^hsa.*\\.txt$", full.names = TRUE) # Initialize an empty list to store the results all_results <- list() # Process each KEGG file for (file in kegg_files) { result <- process_kegg_file(file, zscore_CT_male) all_results[[file]] <- result } # Combine all results into one data frame by merging them on Sample_ID final_results_CT_male <- Reduce(function(x, y) merge(x, y, by = "Sample_ID", all = TRUE), all_results) # Write the final results to a file write.table(final_results_CT_male, "Pathway_zscore_CT_male_summary.txt", sep = "\t", quote = FALSE, row.names = FALSE) ########################################################################################################################################################### # Transpose the file to count 1st and 99th percentile of the Z score distributions in individual pathway final_results_CT_male_t <- as.data.frame(t(final_results_CT_male[,-1])) colnames(final_results_CT_male_t) <- final_results_CT_male$Sample_ID # Convert the row names to a new column named "Pathway_ID" final_results_CT_male_t$Pathway_ID <- rownames(final_results_CT_male_t) # Reorder the columns so that "Pathway_ID" comes first final_results_CT_male_t <- final_results_CT_male_t[, c("Pathway_ID", names(final_results_CT_male_t)[-ncol(final_results_CT_male_t)])] # Reset the row names as they are no longer needed rownames(final_results_CT_male_t) <- NULL # Print the table using DT datatable(final_results_CT_male_t, options = list(pageLength = 5, autoWidth = TRUE))

# Function to calculate percentiles calc_percentiles <- function(x) { lower <- quantile(x, probs = 0.01, na.rm = TRUE) upper <- quantile(x, probs = 0.99, na.rm = TRUE) return(c(lower, upper)) } # Apply the function to each row percentiles <- t(apply(final_results_CT_male_t[,-1], 1, calc_percentiles)) # Add the percentiles to the data frame final_results_CT_male_t_percentile <- cbind(final_results_CT_male_t, lower_1st_percentile = percentiles[, 1], upper_99th_percentile = percentiles[, 2]) # Define a dataframe with thresold values for each gene within the control sample zscored_threshold_male <- final_results_CT_male_t_percentile[,c("Pathway_ID", "lower_1st_percentile", "upper_99th_percentile")] # Write the z-scored and 5th and 95th Percentiles for Each Gene to a text file write.table(zscored_threshold_male, "zscored_percentiles_pathway_data_DLPFC_contorl_male.txt", sep="\t", quote=FALSE, row.names=FALSE) # Print the table using DT datatable(zscored_threshold_male, options = list(pageLength = 5, autoWidth = TRUE))
############################################################################################################################################################ # Plot the data # Set rownames to Sample_ID and remove Sample_ID column rownames(final_results_CT_male) <- final_results_CT_male$Sample_ID final_results_CT_male <- final_results_CT_male[, -1] # Remove columns with NA values final_results_CT_male <- final_results_CT_male[, colSums(is.na(final_results_CT_male)) == 0] # Set up the color scale using RColorBrewer color_scale <- colorRampPalette(brewer.pal(9, "RdYlGn")) # Plot the heatmap using heatmap.2 heatmap.2( as.matrix(final_results_CT_male), Rowv = TRUE, Colv = TRUE, col = color_scale, scale = "column", # Scale rows (genes) to Z-scores trace = "none", # Turn off row and column annotations margins = c(20, 20), # Set margins for row and column labels #col = cm.colors(256), # Set the color map (256 colors) key = TRUE, # Include a color key keysize = 1, # Size of the color key cexCol = 1, # Set column label size cexRow = 0.8, # Set row label size dendrogram = "both", # Show both row and column dendrograms main="Pathway-specific Z score for CT Individuals (male)" )
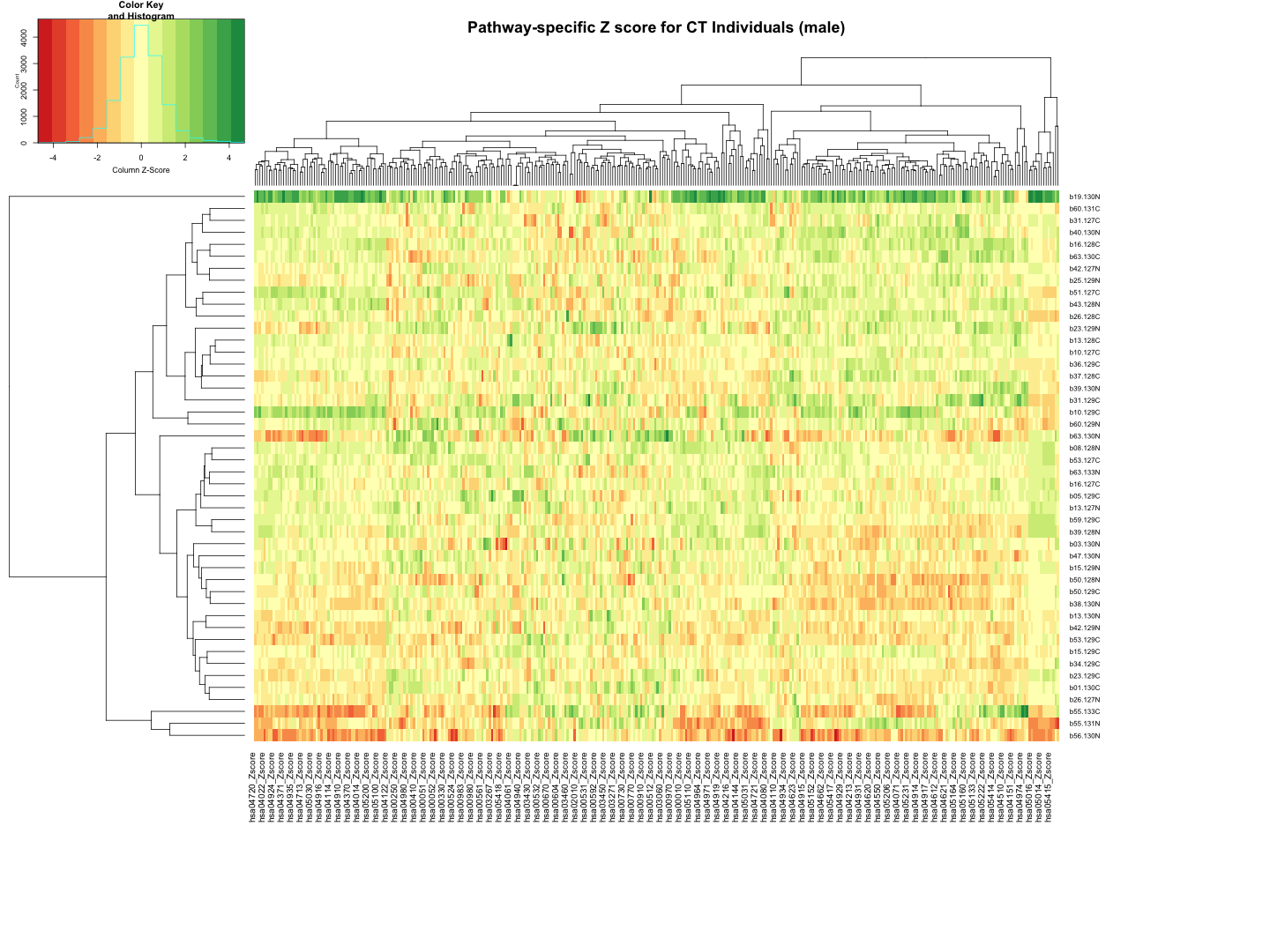
Identfy significant pathways for AD and AsymAD (male and female) using the Z-score thresold (percentile upper and lower bound) from control samples
library(ggplot2) library(tidyr) library(ggdendro) ############################################################################################################################################################################ ## AD_female # Merge both the data frame by Pathway_ID merged_data_female_AD <- merge(zscored_threshold_female, final_results_AD_female_t, by = "Pathway_ID") # Initialize an empty data frame to store results sig_pathway_AD_female <- data.frame(Pathway_ID = merged_data_female_AD$Pathway_ID) # Iterate over each column in final_results_AD_fefemale_t (excluding the Pathway_ID) for (col_name in colnames(final_results_AD_female_t)[-1]) { # Apply the conditions and store the actual value or 0 in a new column in sig_pathway_AD_female sig_pathway_AD_female[[col_name]] <- ifelse( merged_data_female_AD[[col_name]] < merged_data_female_AD$lower_1st_percentile, merged_data_female_AD[[col_name]], # Retrieve the value if it's less than the lower 1st percentile ifelse( merged_data_female_AD[[col_name]] > merged_data_female_AD$upper_99th_percentile, merged_data_female_AD[[col_name]], # Retrieve the value if it's greater than the upper 99th percentile 0 # Otherwise, set to 0 ) ) } # Select only numeric columns (assuming all columns except Pathway_ID are numeric) numeric_columns <- sig_pathway_AD_female[, sapply(sig_pathway_AD_female, is.numeric)] # Calculate UP_count (count of positive values) for each row sig_pathway_AD_female$UP_Count <- rowSums(numeric_columns > 0) # Calculate DOWN_count (count of negative values) for each row sig_pathway_AD_female$DOWN_Count <- rowSums(numeric_columns < 0) # Write the output to a .txt file write.table(sig_pathway_AD_female, "sig_pathway_AD_female.txt", sep = "\t", quote = FALSE, row.names = FALSE) # Print the table using DT datatable(sig_pathway_AD_female, options = list(pageLength = 5, autoWidth = TRUE))

################################################################################################################################################### # Plot the data # Remove the UP_Count and DOWN_Count columns heatmap_data_AD_female <- sig_pathway_AD_female[, !(colnames(sig_pathway_AD_female) %in% c("UP_Count", "DOWN_Count"))] # Set the row names to Pathway_ID and remove the Pathway_ID column row.names(heatmap_data_AD_female) <- sig_pathway_AD_female$Pathway_ID heatmap_data_AD_female$Pathway_ID <- NULL # Check and handle NA/NaN/Inf values heatmap_data_AD_female[is.na(heatmap_data_AD_female)] <- 0 # Replace NA values with 0 heatmap_data_AD_female[is.infinite(heatmap_data_AD_female)] <- 0 # Replace Inf/-Inf values with 0
## Error in is.infinite(heatmap_data_AD_female): default method not implemented for type 'list'
# Convert the data to numeric type (if it's not already) heatmap_data_AD_female <- as.matrix(heatmap_data_AD_female) # Define the color palette color_palette <- colorRampPalette(c("red", "white", "blue"))(100) # Generate the heatmap out_heatmap_AD_female <- heatmap.2( heatmap_data_AD_female, # Data matrix Rowv = TRUE, # Row dendrogram Colv = TRUE, # Column dendrogram col = color_palette, # Color palette scale = "row", # No scaling trace = "none", # Turn off trace lines margins = c(10, 10), # Set margins for labels key = TRUE, # Include color key key.title = "Z-Score", # Key title keysize = 1.5, # Size of the key cexRow = 0.8, # Size of row labels cexCol = 0.8, # Size of column labels dendrogram = "both", # Show both row and column dendrograms main = "Pathway Specific Z-Scores Distribution (AD female)" # Main title )
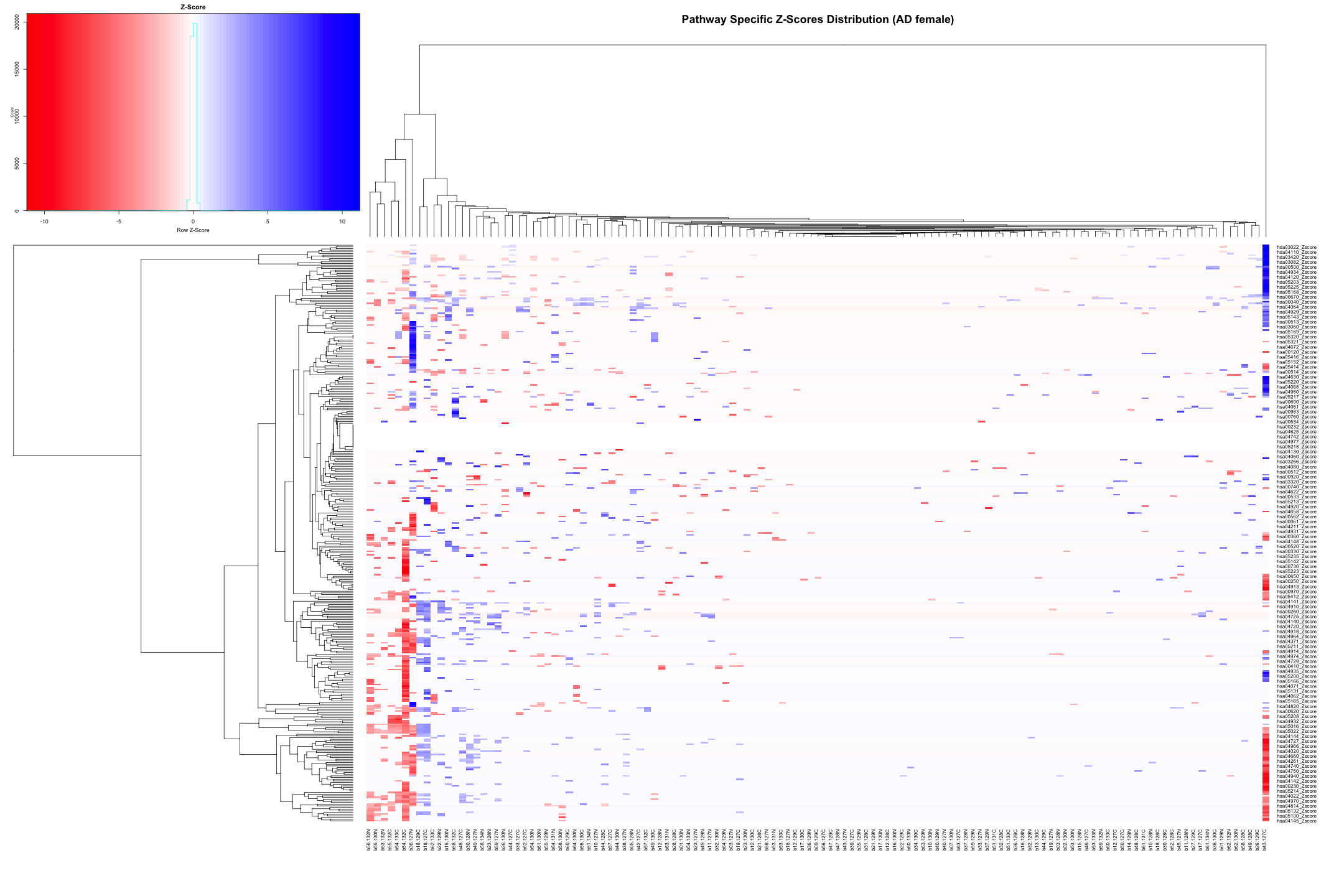
# Plot the dendogram ggdendrogram(out_heatmap_AD_female$colDendrogram, theme_dendro = FALSE)
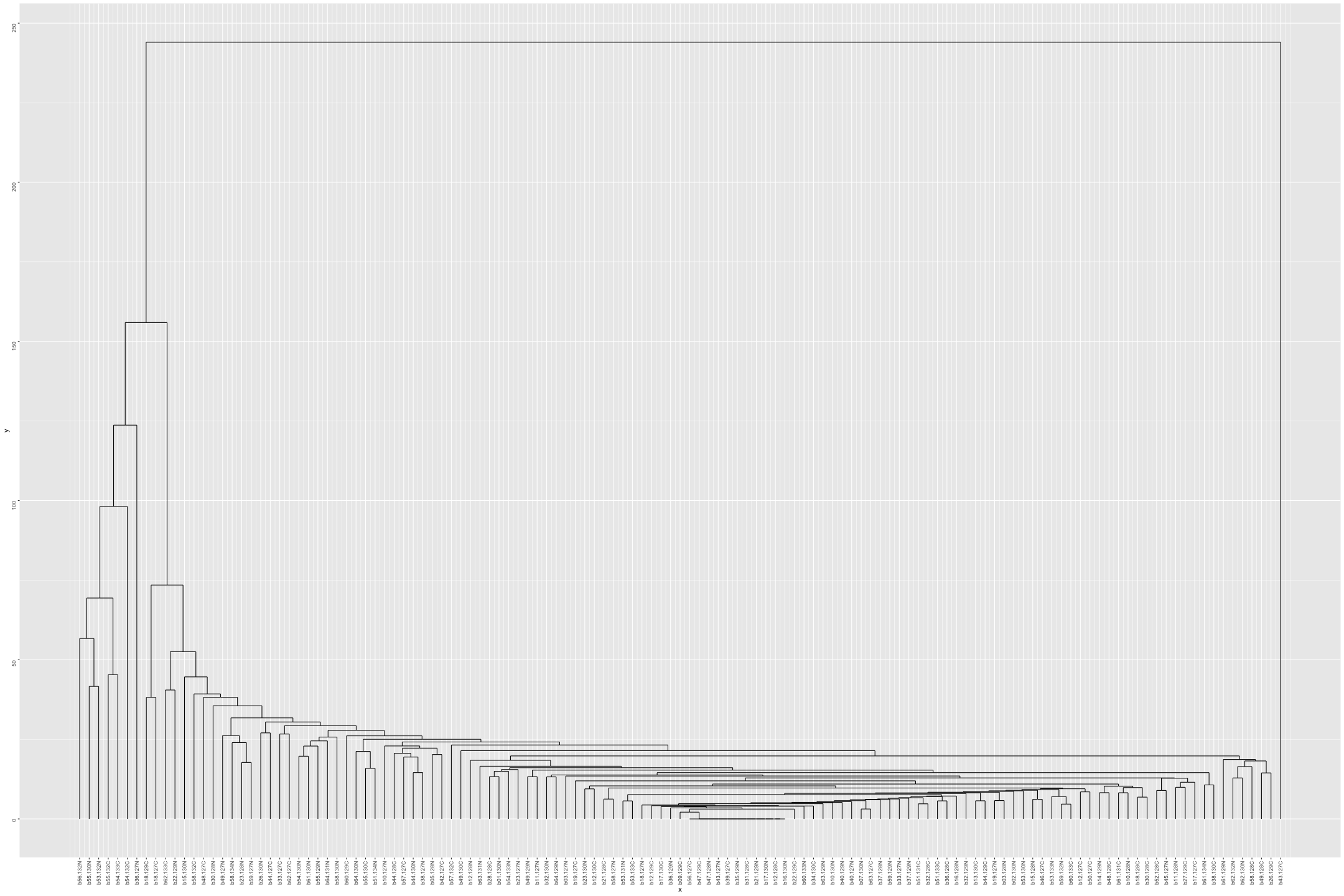
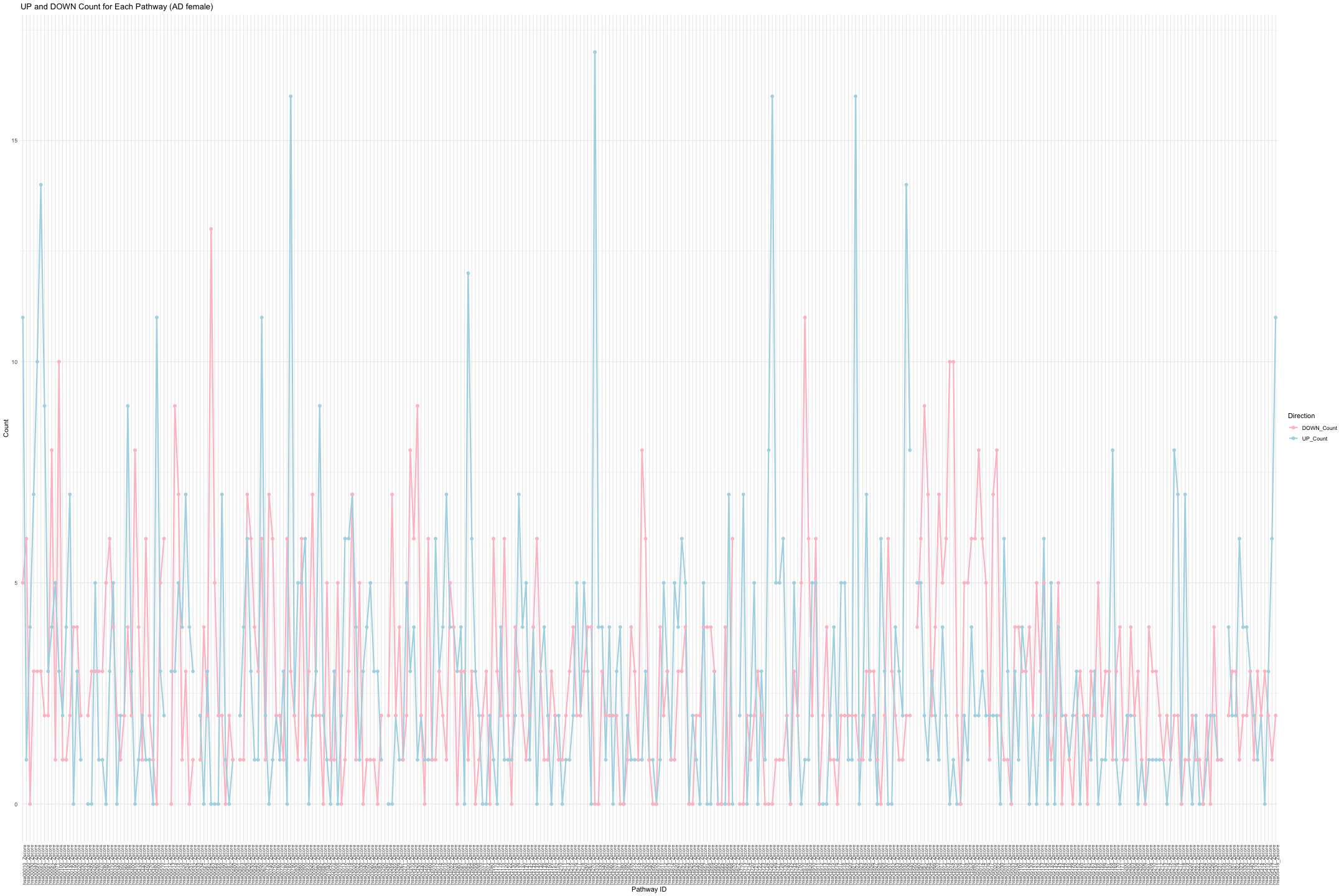
# Convert data from wide to long format long_sig_pathway_AD_female <- gather(sig_pathway_AD_female, key = "Direction", value = "Count", UP_Count, DOWN_Count) # Create the line plot ggplot(long_sig_pathway_AD_female, aes(x = Pathway_ID, y = Count, color = Direction, group = Direction)) + geom_line(size = 1) + # Add lines between points geom_point(size = 2) + # Add points at each Pathway_ID theme_minimal() + labs(title = "UP and DOWN Count for Each Pathway (AD female)", x = "Pathway ID", y = "Count") + theme(axis.text.x = element_text(angle = 90, hjust = 1, size = 8)) + # Rotate x-axis labels if needed scale_color_manual(values = c("UP_Count" = "lightblue", "DOWN_Count" = "pink")) # Customize colors
## Warning: Using `size` aesthetic for lines was deprecated in ggplot2 3.4.0. ## ℹ Please use `linewidth` instead. ## This warning is displayed once every 8 hours. ## Call `lifecycle::last_lifecycle_warnings()` to see where this warning was ## generated.
## Warning: Removed 16 rows containing missing values or values outside the scale range ## (`geom_point()`).
############################################################################################################################################################################ ############################################################################################################################################################################ ## AD_male # Merge both the data frame by Pathway_ID merged_data_male_AD <- merge(zscored_threshold_male, final_results_AD_male_t, by = "Pathway_ID") # Initialize an empty data frame to store results sig_pathway_AD_male <- data.frame(Pathway_ID = merged_data_male_AD$Pathway_ID) # Iterate over each column in final_results_AD_female_t (excluding the Pathway_ID) for (col_name in colnames(final_results_AD_male_t)[-1]) { # Apply the conditions and store the actual value or 0 in a new column in sig_pathway_AD_male sig_pathway_AD_male[[col_name]] <- ifelse( merged_data_male_AD[[col_name]] < merged_data_male_AD$lower_1st_percentile, merged_data_male_AD[[col_name]], # Retrieve the value if it's less than the lower 1st percentile ifelse( merged_data_male_AD[[col_name]] > merged_data_male_AD$upper_99th_percentile, merged_data_male_AD[[col_name]], # Retrieve the value if it's greater than the upper 99th percentile 0 # Otherwise, set to 0 ) ) } # Select only numeric columns (assuming all columns except Pathway_ID are numeric) numeric_columns <- sig_pathway_AD_male[, sapply(sig_pathway_AD_male, is.numeric)] # Calculate UP_count (count of positive values) for each row sig_pathway_AD_male$UP_Count <- rowSums(numeric_columns > 0) # Calculate DOWN_count (count of negative values) for each row sig_pathway_AD_male$DOWN_Count <- rowSums(numeric_columns < 0) # Write the output to a .txt file write.table(sig_pathway_AD_male, "sig_pathway_AD_male.txt", sep = "\t", quote = FALSE, row.names = FALSE) # Print the table using DT datatable(sig_pathway_AD_male, options = list(pageLength = 5, autoWidth = TRUE))
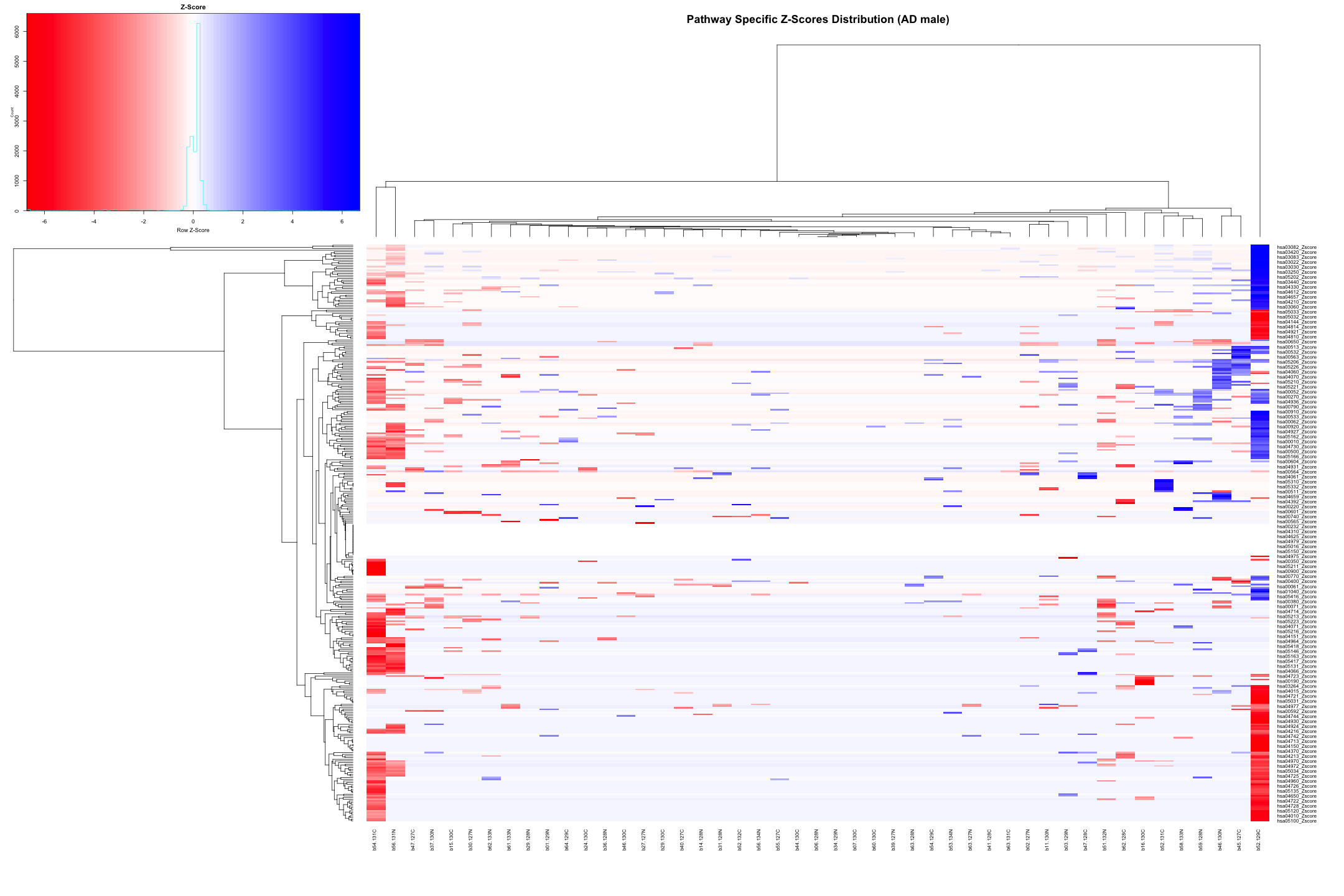
################################################################################################################################################### # Plot the data # Remove the UP_Count and DOWN_Count columns heatmap_data_AD_male <- sig_pathway_AD_male[, !(colnames(sig_pathway_AD_male) %in% c("UP_Count", "DOWN_Count"))] # Set the row names to Pathway_ID and remove the Pathway_ID column row.names(heatmap_data_AD_male) <- sig_pathway_AD_male$Pathway_ID heatmap_data_AD_male$Pathway_ID <- NULL # Check and handle NA/NaN/Inf values heatmap_data_AD_male[is.na(heatmap_data_AD_male)] <- 0 # Replace NA values with 0 heatmap_data_AD_male[is.infinite(heatmap_data_AD_male)] <- 0 # Replace Inf/-Inf values with 0
## Error in is.infinite(heatmap_data_AD_male): default method not implemented for type 'list'
# Convert the data to numeric type (if it's not already) heatmap_data_AD_male <- as.matrix(heatmap_data_AD_male) # Define the color palette color_palette <- colorRampPalette(c("red", "white", "blue"))(100) # Generate the heatmap out_heatmap_AD_male <- heatmap.2( heatmap_data_AD_male, # Data matrix Rowv = TRUE, # Row dendrogram Colv = TRUE, # Column dendrogram col = color_palette, # Color palette scale = "row", # No scaling trace = "none", # Turn off trace lines margins = c(10, 10), # Set margins for labels key = TRUE, # Include color key key.title = "Z-Score", # Key title keysize = 1.5, # Size of the key cexRow = 0.8, # Size of row labels cexCol = 0.8, # Size of column labels dendrogram = "both", # Show both row and column dendrograms main = "Pathway Specific Z-Scores Distribution (AD male)" # Main title )
# Plot the dendogram ggdendrogram(out_heatmap_AD_male$colDendrogram, theme_dendro = FALSE)
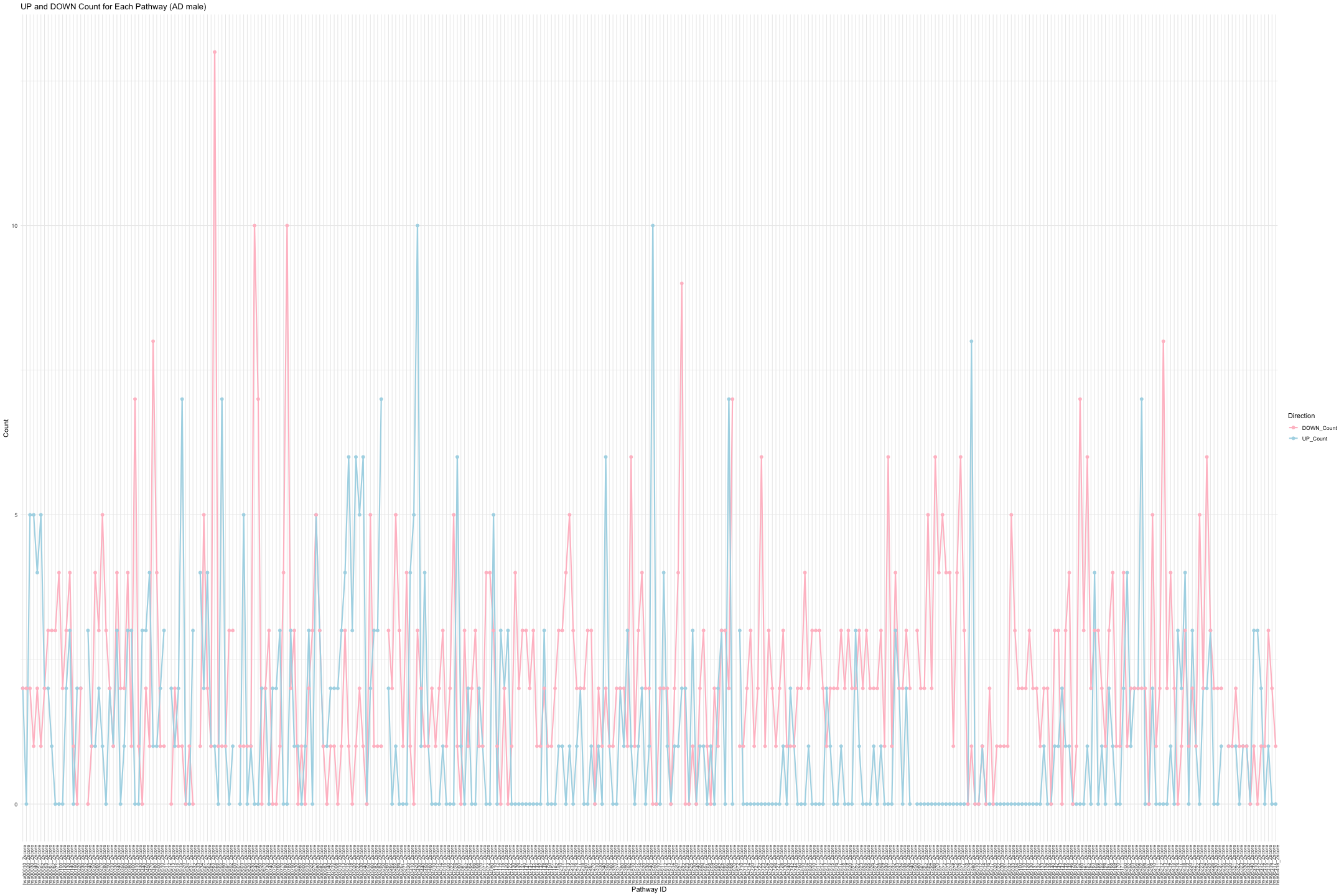
# Convert data from wide to long format long_sig_pathway_AD_male <- gather(sig_pathway_AD_male, key = "Direction", value = "Count", UP_Count, DOWN_Count) # Create the line plot ggplot(long_sig_pathway_AD_male, aes(x = Pathway_ID, y = Count, color = Direction, group = Direction)) + geom_line(size = 1) + # Add lines between points geom_point(size = 2) + # Add points at each Pathway_ID theme_minimal() + labs(title = "UP and DOWN Count for Each Pathway (AD male)", x = "Pathway ID", y = "Count") + theme(axis.text.x = element_text(angle = 90, hjust = 1, size = 8)) + # Rotate x-axis labels if needed scale_color_manual(values = c("UP_Count" = "lightblue", "DOWN_Count" = "pink")) # Customize colors
## Warning: Removed 16 rows containing missing values or values outside the scale range ## (`geom_point()`).
############################################################################################################################################################################ ############################################################################################################################################################################ ## AsymAD_female # Merge both the data frame by Pathway_ID merged_data_female_AsymAD <- merge(zscored_threshold_female, final_results_AsymAD_female_t, by = "Pathway_ID") # Initialize an empty data frame to store results sig_pathway_AsymAD_female <- data.frame(Pathway_ID = merged_data_female_AsymAD$Pathway_ID) # Iterate over each column in B (excluding the Pathway_ID) #for (col_name in colnames(final_results_AsymAD_female_t)[-1]) { # Apply the conditions and store the result in a new column in Output #sig_pathway_AsymAD_female[[col_name]] <- ifelse(merged_data_female_AsymAD[[col_name]] < merged_data_female_AsymAD$lower_1st_percentile, #"DOWN", #ifelse(merged_data_female_AsymAD[[col_name]] > merged_data_female_AsymAD$upper_99th_percentile, #"UP", #"0")) #} # Add a column to count the number of "UP" occurrences in each row #sig_pathway_AsymAD_female$UP_Count <- rowSums(sig_pathway_AsymAD_female == "UP") # Add a column to count the number of "DOWN" occurrences in each row #sig_pathway_AsymAD_female$DOWN_Count <- rowSums(sig_pathway_AsymAD_female == "DOWN") # Iterate over each column in final_results_AsymAD_female_t (excluding the Pathway_ID) for (col_name in colnames(final_results_AsymAD_female_t)[-1]) { # Apply the conditions and store the actual value or 0 in a new column in sig_pathway_AsymAD_female sig_pathway_AsymAD_female[[col_name]] <- ifelse( merged_data_female_AsymAD[[col_name]] < merged_data_female_AsymAD$lower_1st_percentile, merged_data_female_AsymAD[[col_name]], # Retrieve the value if it's less than the lower 1st percentile ifelse( merged_data_female_AsymAD[[col_name]] > merged_data_female_AsymAD$upper_99th_percentile, merged_data_female_AsymAD[[col_name]], # Retrieve the value if it's greater than the upper 99th percentile 0 # Otherwise, set to 0 ) ) } # Select only numeric columns (assuming all columns except Pathway_ID are numeric) numeric_columns <- sig_pathway_AsymAD_female[, sapply(sig_pathway_AsymAD_female, is.numeric)] # Calculate UP_count (count of positive values) for each row sig_pathway_AsymAD_female$UP_Count <- rowSums(numeric_columns > 0) # Calculate DOWN_count (count of negative values) for each row sig_pathway_AsymAD_female$DOWN_Count <- rowSums(numeric_columns < 0) # Write the output to a .txt file write.table(sig_pathway_AsymAD_female, "sig_pathway_AsymAD_female.txt", sep = "\t", quote = FALSE, row.names = FALSE) # Print the table using DT datatable(sig_pathway_AsymAD_female, options = list(pageLength = 5, autoWidth = TRUE))

################################################################################################################################################### # Plot the data # Remove the UP_Count and DOWN_Count columns heatmap_data_AsymAD_female <- sig_pathway_AsymAD_female[, !(colnames(sig_pathway_AsymAD_female) %in% c("UP_Count", "DOWN_Count"))] # Set the row names to Pathway_ID and remove the Pathway_ID column row.names(heatmap_data_AsymAD_female) <- sig_pathway_AsymAD_female$Pathway_ID heatmap_data_AsymAD_female$Pathway_ID <- NULL # Check and handle NA/NaN/Inf values heatmap_data_AsymAD_female[is.na(heatmap_data_AsymAD_female)] <- 0 # Replace NA values with 0 heatmap_data_AsymAD_female[is.infinite(heatmap_data_AsymAD_female)] <- 0 # Replace Inf/-Inf values with 0
## Error in is.infinite(heatmap_data_AsymAD_female): default method not implemented for type 'list'
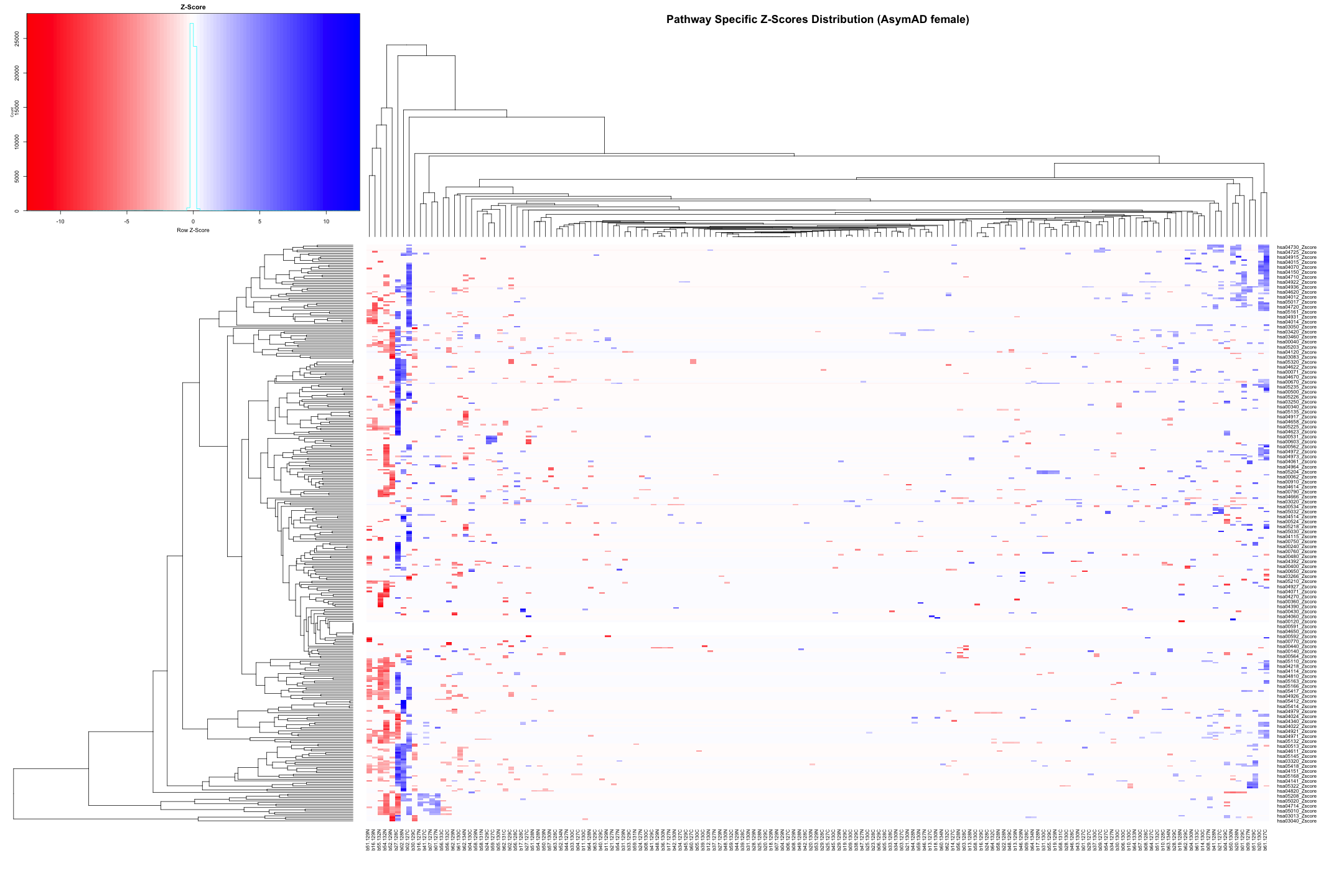
# Convert the data to numeric type (if it's not already) heatmap_data_AsymAD_female <- as.matrix(heatmap_data_AsymAD_female) # Define the color palette color_palette <- colorRampPalette(c("red", "white", "blue"))(100) # Generate the heatmap out_heatmap_AsymAD_female <- heatmap.2( heatmap_data_AsymAD_female, # Data matrix Rowv = TRUE, # Row dendrogram Colv = TRUE, # Column dendrogram col = color_palette, # Color palette scale = "row", # No scaling trace = "none", # Turn off trace lines margins = c(10, 10), # Set margins for labels key = TRUE, # Include color key key.title = "Z-Score", # Key title keysize = 1.5, # Size of the key cexRow = 0.8, # Size of row labels cexCol = 0.8, # Size of column labels dendrogram = "both", # Show both row and column dendrograms main = "Pathway Specific Z-Scores Distribution (AsymAD female)" # Main title )
# Plot the dendogram ggdendrogram(out_heatmap_AsymAD_female$colDendrogram, theme_dendro = TRUE)
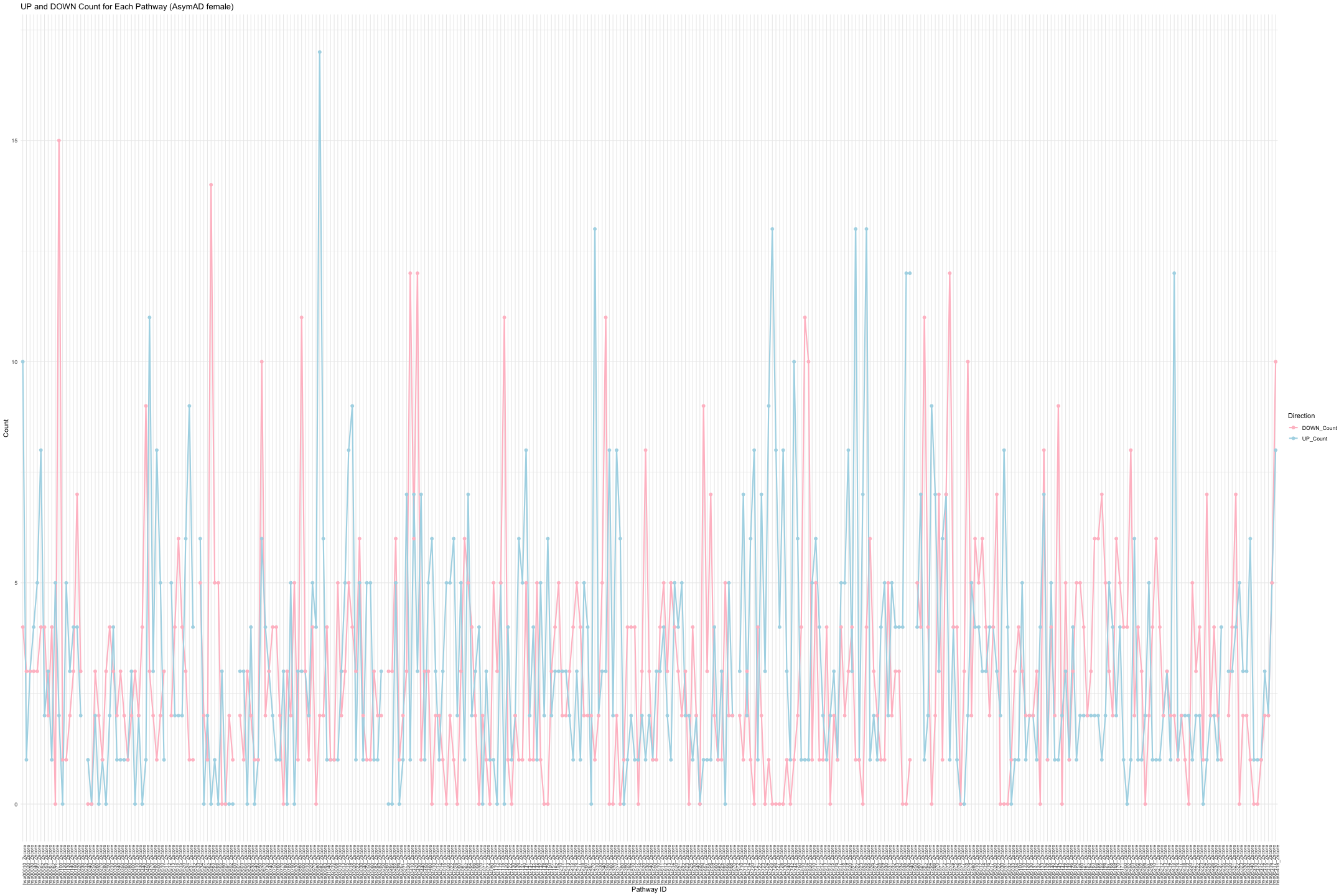
# Convert data from wide to long format long_sig_pathway_AsymAD_female <- gather(sig_pathway_AsymAD_female, key = "Direction", value = "Count", UP_Count, DOWN_Count) # Create the bar plot ggplot(long_sig_pathway_AsymAD_female, aes(x = Pathway_ID, y = Count, color = Direction, group = Direction)) + geom_line(size = 1) + # Add lines between points geom_point(size = 2) + # Add points at each Pathway_ID theme_minimal() + labs(title = "UP and DOWN Count for Each Pathway (AsymAD female)", x = "Pathway ID", y = "Count") + theme(axis.text.x = element_text(angle = 90, hjust = 1, size = 8)) + # Rotate x-axis labels if needed scale_color_manual(values = c("UP_Count" = "lightblue", "DOWN_Count" = "pink")) # Customize colors
## Warning: Removed 16 rows containing missing values or values outside the scale range ## (`geom_point()`).
############################################################################################################################################################################ ############################################################################################################################################################################ ## AsymAD_male # Merge both the data frame by Pathway_ID merged_data_male_AsymAD <- merge(zscored_threshold_male, final_results_AsymAD_male_t, by = "Pathway_ID") # Initialize an empty data frame to store results sig_pathway_AsymAD_male <- data.frame(Pathway_ID = merged_data_male_AsymAD$Pathway_ID) # Iterate over each column in final_results_AsymAD_female_t (excluding the Pathway_ID) for (col_name in colnames(final_results_AsymAD_male_t)[-1]) { # Apply the conditions and store the actual value or 0 in a new column in sig_pathway_AsymAD_male sig_pathway_AsymAD_male[[col_name]] <- ifelse( merged_data_male_AsymAD[[col_name]] < merged_data_male_AsymAD$lower_1st_percentile, merged_data_male_AsymAD[[col_name]], # Retrieve the value if it's less than the lower 1st percentile ifelse( merged_data_male_AsymAD[[col_name]] > merged_data_male_AsymAD$upper_99th_percentile, merged_data_male_AsymAD[[col_name]], # Retrieve the value if it's greater than the upper 99th percentile 0 # Otherwise, set to 0 ) ) } # Select only numeric columns (assuming all columns except Pathway_ID are numeric) numeric_columns <- sig_pathway_AsymAD_male[, sapply(sig_pathway_AsymAD_male, is.numeric)] # Calculate UP_count (count of positive values) for each row sig_pathway_AsymAD_male$UP_Count <- rowSums(numeric_columns > 0) # Calculate DOWN_count (count of negative values) for each row sig_pathway_AsymAD_male$DOWN_Count <- rowSums(numeric_columns < 0) # Write the output to a .txt file write.table(sig_pathway_AsymAD_male, "sig_pathway_AsymAD_male.txt", sep = "\t", quote = FALSE, row.names = FALSE) # Print the table using DT datatable(sig_pathway_AsymAD_male, options = list(pageLength = 5, autoWidth = TRUE))

################################################################################################################################################### # Plot the data # Remove the UP_Count and DOWN_Count columns heatmap_data_AsymAD_male <- sig_pathway_AsymAD_male[, !(colnames(sig_pathway_AsymAD_male) %in% c("UP_Count", "DOWN_Count"))] # Set the row names to Pathway_ID and remove the Pathway_ID column row.names(heatmap_data_AsymAD_male) <- sig_pathway_AsymAD_male$Pathway_ID heatmap_data_AsymAD_male$Pathway_ID <- NULL # Check and handle NA/NaN/Inf values heatmap_data_AsymAD_male[is.na(heatmap_data_AsymAD_male)] <- 0 # Replace NA values with 0 heatmap_data_AsymAD_male[is.infinite(heatmap_data_AsymAD_male)] <- 0 # Replace Inf/-Inf values with 0
## Error in is.infinite(heatmap_data_AsymAD_male): default method not implemented for type 'list'
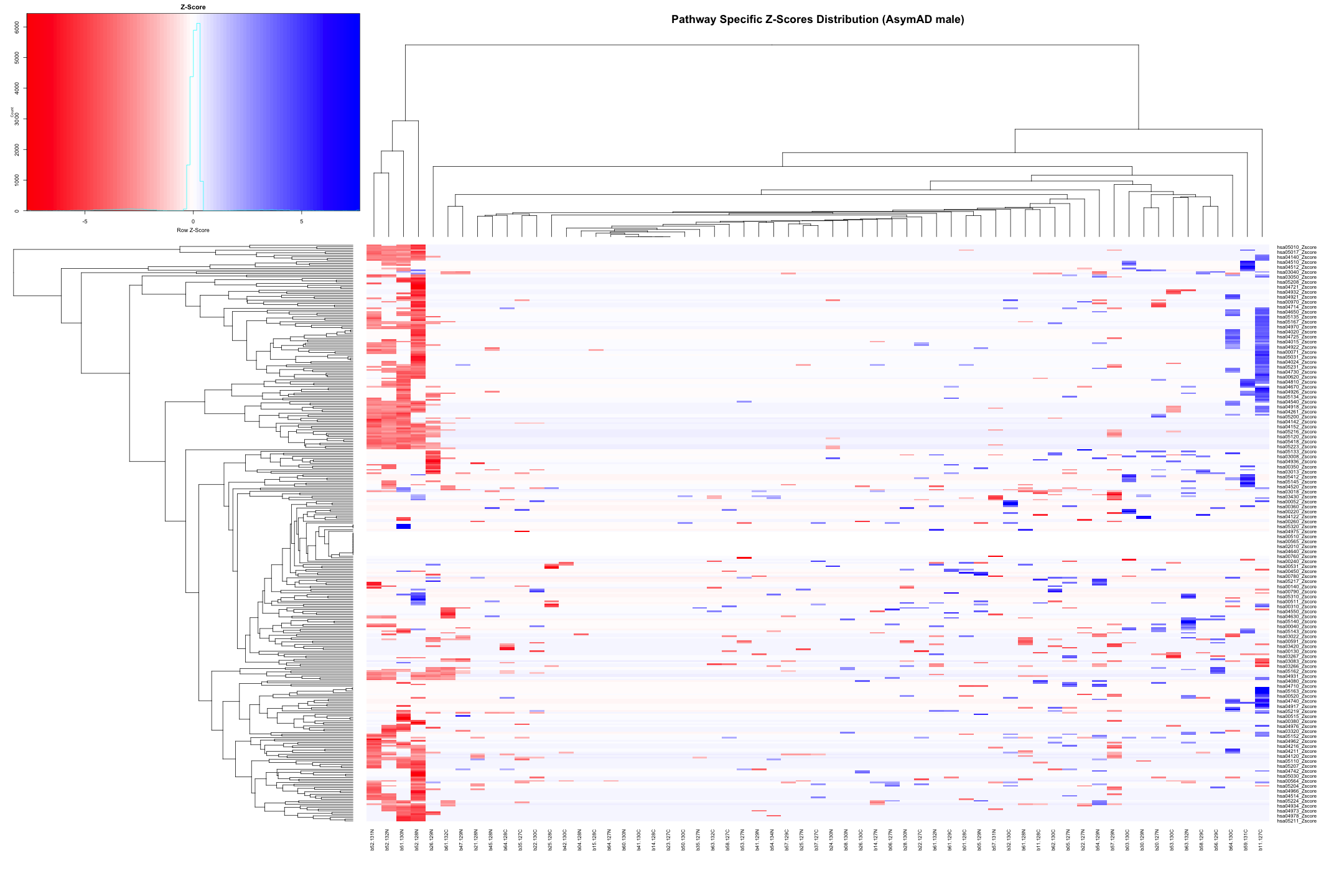
# Convert the data to numeric type (if it's not already) heatmap_data_AsymAD_male <- as.matrix(heatmap_data_AsymAD_male) # Define the color palette color_palette <- colorRampPalette(c("red", "white", "blue"))(100) # Generate the heatmap out_heatmap_AsymAD_male <- heatmap.2( heatmap_data_AsymAD_male, # Data matrix Rowv = TRUE, # Row dendrogram Colv = TRUE, # Column dendrogram col = color_palette, # Color palette scale = "row", # No scaling trace = "none", # Turn off trace lines margins = c(10, 10), # Set margins for labels key = TRUE, # Include color key key.title = "Z-Score", # Key title keysize = 1.5, # Size of the key cexRow = 0.8, # Size of row labels cexCol = 0.8, # Size of column labels dendrogram = "both", # Show both row and column dendrograms main = "Pathway Specific Z-Scores Distribution (AsymAD male)" # Main title )
# Plot the dendogram ggdendrogram(out_heatmap_AsymAD_male$colDendrogram, theme_dendro = FALSE)
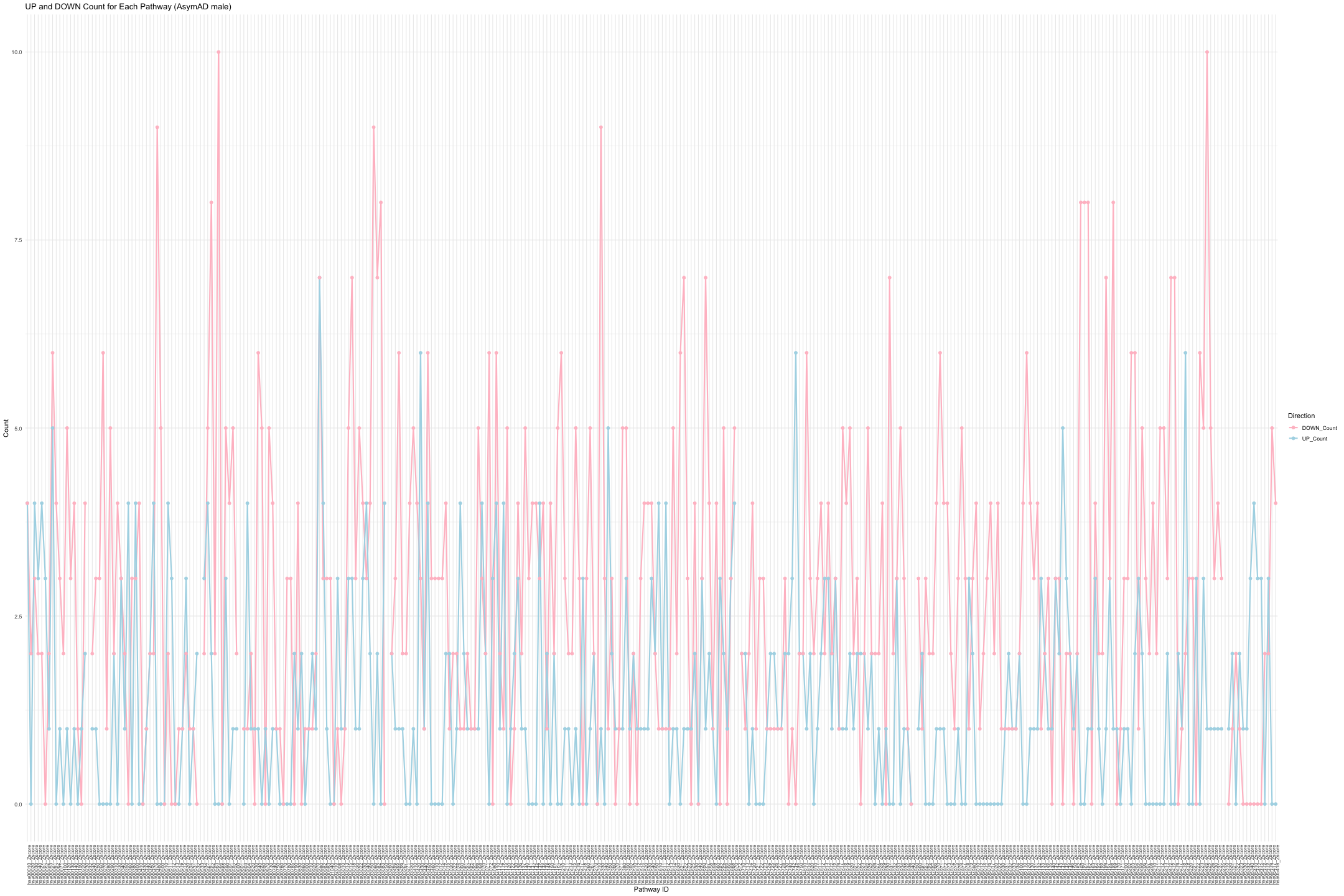
# Convert data from wide to long format long_sig_pathway_AsymAD_male <- gather(sig_pathway_AsymAD_male, key = "Direction", value = "Count", UP_Count, DOWN_Count) # Create the line plot ggplot(long_sig_pathway_AsymAD_male, aes(x = Pathway_ID, y = Count, color = Direction, group = Direction)) + geom_line(size = 1) + # AsymADd lines between points geom_point(size = 2) + # Add points at each Pathway_ID theme_minimal() + labs(title = "UP and DOWN Count for Each Pathway (AsymAD male)", x = "Pathway ID", y = "Count") + theme(axis.text.x = element_text(angle = 90, hjust = 1, size = 8)) + # Rotate x-axis labels if needed scale_color_manual(values = c("UP_Count" = "lightblue", "DOWN_Count" = "pink")) # Customize colors
## Warning: Removed 14 rows containing missing values or values outside the scale range ## (`geom_point()`).
############################################################################################################################################################################ ############################################################################################################################################################################ ## CT_female # Merge both data frames by Pathway_ID merged_data_female_CT <- merge(zscored_threshold_female, final_results_CT_female_t, by = "Pathway_ID", all.x = TRUE) # Initialize an empty data frame to store results sig_pathway_CT_female <- data.frame(Pathway_ID = merged_data_female_CT$Pathway_ID) # Iterate over each column in final_results_CT_female_t (excluding the Pathway_ID) for (col_name in colnames(final_results_CT_female_t)[-1]) { # Apply the conditions and store the actual value or 0 in a new column in sig_pathway_CT_female sig_pathway_CT_female[[col_name]] <- ifelse( merged_data_female_CT[[col_name]] < merged_data_female_CT$lower_1st_percentile, merged_data_female_CT[[col_name]], # Retrieve the value if it's less than the lower 1st percentile ifelse( merged_data_female_CT[[col_name]] > merged_data_female_CT$upper_99th_percentile, merged_data_female_CT[[col_name]], # Retrieve the value if it's greater than the upper 99th percentile 0 # Otherwise, set to 0 ) ) } # Select only numeric columns (assuming all columns except Pathway_ID are numeric) numeric_columns <- sig_pathway_CT_female[, sapply(sig_pathway_CT_female, is.numeric)] # Calculate UP_count (count of positive values) for each row sig_pathway_CT_female$UP_Count <- rowSums(numeric_columns > 0) # Calculate DOWN_count (count of negative values) for each row sig_pathway_CT_female$DOWN_Count <- rowSums(numeric_columns < 0) # Write the output to a .txt file write.table(sig_pathway_CT_female, "sig_pathway_CT_female.txt", sep = "\t", quote = FALSE, row.names = FALSE) # Print the table using DT datatable(sig_pathway_CT_female, options = list(pageLength = 5, autoWidth = TRUE))

################################################################################################################################################### # Plot the data # Remove the UP_Count and DOWN_Count columns heatmap_data_CT_female <- sig_pathway_CT_female[, !(colnames(sig_pathway_CT_female) %in% c("UP_Count", "DOWN_Count"))] # Set the row names to Pathway_ID and remove the Pathway_ID column row.names(heatmap_data_CT_female) <- sig_pathway_CT_female$Pathway_ID heatmap_data_CT_female$Pathway_ID <- NULL # Check and handle NA/NaN/Inf values heatmap_data_CT_female[is.na(heatmap_data_CT_female)] <- 0 # Replace NA values with 0 heatmap_data_CT_female[is.infinite(heatmap_data_CT_female)] <- 0 # Replace Inf/-Inf values with 0
## Error in is.infinite(heatmap_data_CT_female): default method not implemented for type 'list'
# Convert the data to numeric type (if it's not already) heatmap_data_CT_female <- as.matrix(heatmap_data_CT_female) # Define the color palette color_palette <- colorRampPalette(c("red", "white", "blue"))(100) # Generate the heatmap heatmap.2( heatmap_data_CT_female, # Data matrix Rowv = TRUE, # Row dendrogram Colv = TRUE, # Column dendrogram col = color_palette, # Color palette scale = "row", # No scaling trace = "none", # Turn off trace lines margins = c(10, 10), # Set margins for labels key = TRUE, # Include color key key.title = "Z-Score", # Key title keysize = 1.5, # Size of the key cexRow = 0.8, # Size of row labels cexCol = 0.8, # Size of column labels dendrogram = "both", # Show both row and column dendrograms main = "Pathway Specific Z-Scores Distribution (CT female)" # Main title )
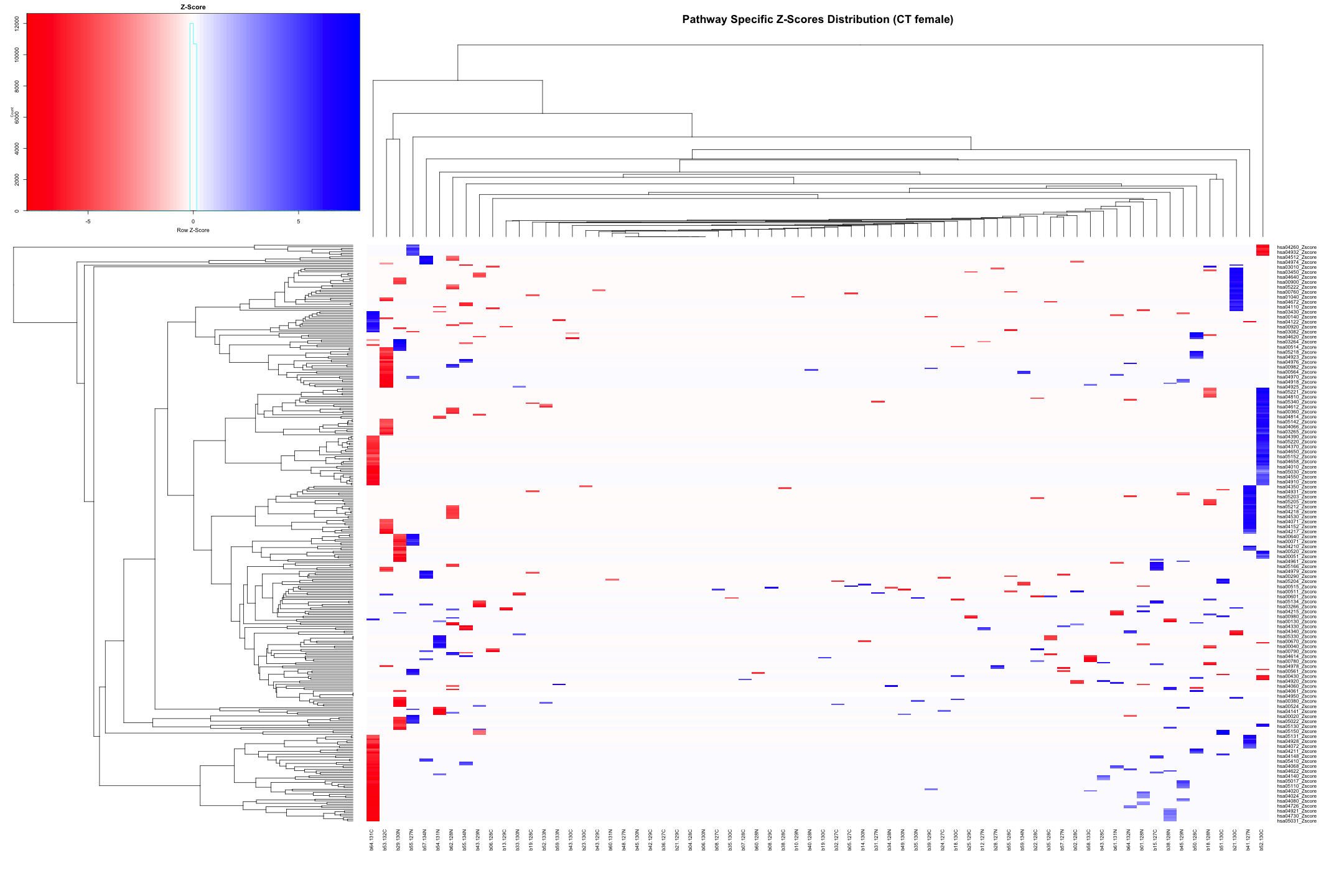
# Convert data from wide to long format long_sig_pathway_CT_female <- gather(sig_pathway_CT_female, key = "Direction", value = "Count", UP_Count, DOWN_Count) # Create the bar plot ggplot(long_sig_pathway_CT_female, aes(x = Pathway_ID, y = Count, fill = Direction)) + geom_bar(stat = "identity", position = "dodge") + theme_minimal() + labs(title = "UP and DOWN Count for Each Pathway (CT female)", x = "Pathway ID", y = "Count") + theme(axis.text.x = element_text(angle = 90, hjust = 1, size = 0.5)) + # Rotate x-axis labels if needed scale_fill_manual(values = c("UP_Count" = "lightblue", "DOWN_Count" = "pink")) # Customize colors
## Warning: Removed 16 rows containing missing values or values outside the scale range ## (`geom_bar()`).

############################################################################################################################################################################ ############################################################################################################################################################################ ## CT_male # Merge both data frames by Pathway_ID merged_data_male_CT <- merge(zscored_threshold_male, final_results_CT_male_t, by = "Pathway_ID", all.x = TRUE) # Initialize an empty data frame to store results sig_pathway_CT_male <- data.frame(Pathway_ID = merged_data_male_CT$Pathway_ID) # Iterate over each column in final_results_CT_female_t (excluding the Pathway_ID) for (col_name in colnames(final_results_CT_male_t)[-1]) { # Apply the conditions and store the actual value or 0 in a new column in sig_pathway_CT_male sig_pathway_CT_male[[col_name]] <- ifelse( merged_data_male_CT[[col_name]] < merged_data_male_CT$lower_1st_percentile, merged_data_male_CT[[col_name]], # Retrieve the value if it's less than the lower 1st percentile ifelse( merged_data_male_CT[[col_name]] > merged_data_male_CT$upper_99th_percentile, merged_data_male_CT[[col_name]], # Retrieve the value if it's greater than the upper 99th percentile 0 # Otherwise, set to 0 ) ) } # Select only numeric columns (assuming all columns except Pathway_ID are numeric) numeric_columns <- sig_pathway_CT_male[, sapply(sig_pathway_CT_male, is.numeric)] # Calculate UP_count (count of positive values) for each row sig_pathway_CT_male$UP_Count <- rowSums(numeric_columns > 0) # Calculate DOWN_count (count of negative values) for each row sig_pathway_CT_male$DOWN_Count <- rowSums(numeric_columns < 0) # Write the output to a .txt file write.table(sig_pathway_CT_male, "sig_pathway_CT_male.txt", sep = "\t", quote = FALSE, row.names = FALSE) # Print the table using DT datatable(sig_pathway_CT_male, options = list(pageLength = 5, autoWidth = TRUE))

################################################################################################################################################### # Plot the data # Remove the UP_Count and DOWN_Count columns heatmap_data_CT_male <- sig_pathway_CT_male[, !(colnames(sig_pathway_CT_male) %in% c("UP_Count", "DOWN_Count"))] # Set the row names to Pathway_ID and remove the Pathway_ID column row.names(heatmap_data_CT_male) <- sig_pathway_CT_male$Pathway_ID heatmap_data_CT_male$Pathway_ID <- NULL # Check and handle NA/NaN/Inf values heatmap_data_CT_male[is.na(heatmap_data_CT_male)] <- 0 # Replace NA values with 0 heatmap_data_CT_male[is.infinite(heatmap_data_CT_male)] <- 0 # Replace Inf/-Inf values with 0
## Error in is.infinite(heatmap_data_CT_male): default method not implemented for type 'list'
# Convert the data to numeric type (if it's not already) heatmap_data_CT_male <- as.matrix(heatmap_data_CT_male) # Define the color palette color_palette <- colorRampPalette(c("red", "white", "blue"))(100) # Generate the heatmap heatmap.2( heatmap_data_CT_male, # Data matrix Rowv = TRUE, # Row dendrogram Colv = TRUE, # Column dendrogram col = color_palette, # Color palette scale = "row", # No scaling trace = "none", # Turn off trace lines margins = c(10, 10), # Set margins for labels key = TRUE, # Include color key key.title = "Z-Score", # Key title keysize = 1.5, # Size of the key cexRow = 0.8, # Size of row labels cexCol = 0.8, # Size of column labels dendrogram = "both", # Show both row and column dendrograms main = "Pathway Specific Z-Scores Distribution (CT male)" # Main title )
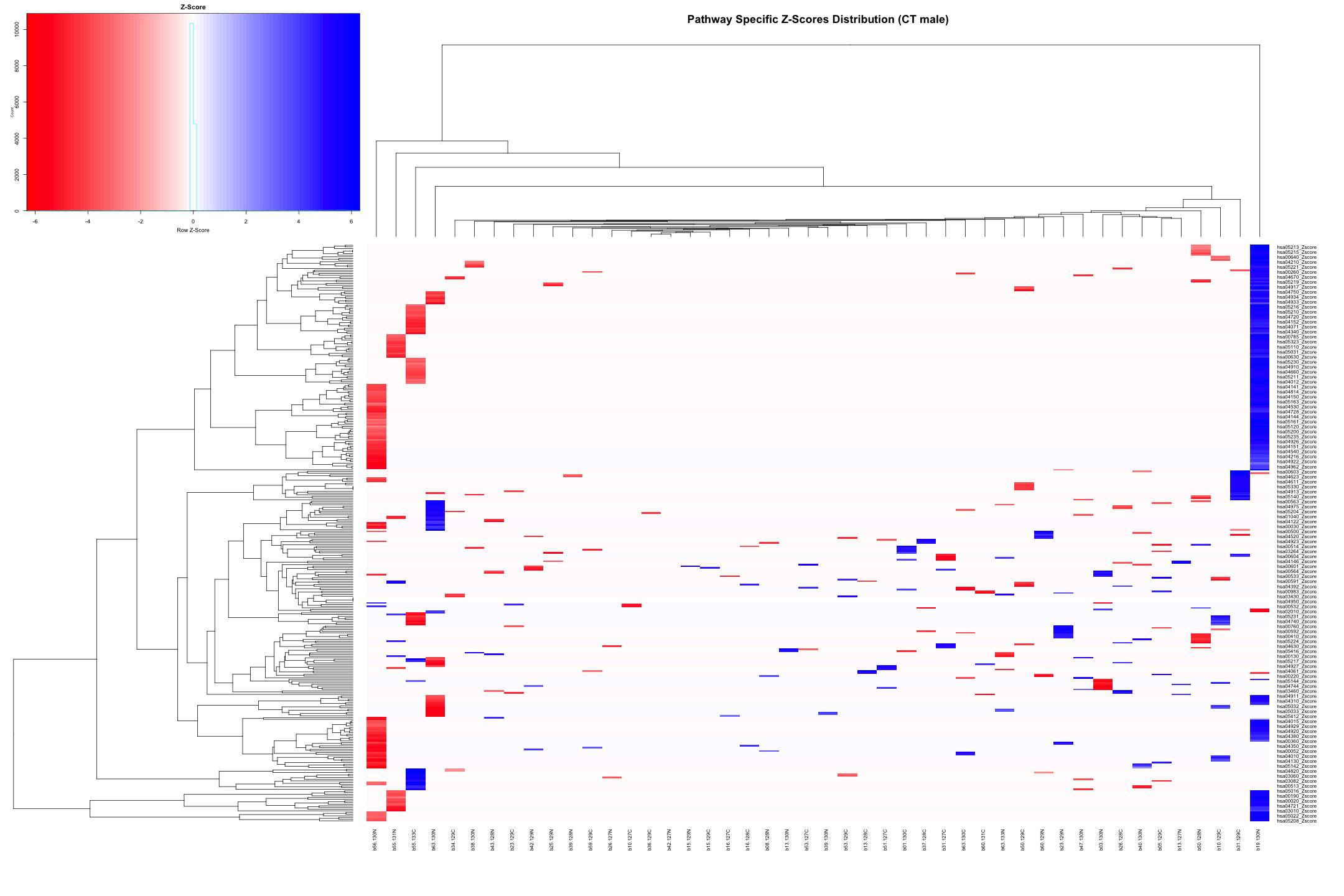
# Convert data from wide to long format long_sig_pathway_CT_male <- gather(sig_pathway_CT_male, key = "Direction", value = "Count", UP_Count, DOWN_Count) # Create the bar plot ggplot(long_sig_pathway_CT_male, aes(x = Pathway_ID, y = Count, fill = Direction)) + geom_bar(stat = "identity", position = "dodge") + theme_minimal() + labs(title = "UP and DOWN Count for Each Pathway (CT male)", x = "Pathway ID", y = "Count") + theme(axis.text.x = element_text(angle = 90, hjust = 1, size = 0.5)) + # Rotate x-axis labels if needed scale_fill_manual(values = c("UP_Count" = "lightblue", "DOWN_Count" = "pink")) # Customize colors
## Warning: Removed 14 rows containing missing values or values outside the scale range ## (`geom_bar()`).

Compare Pathway-specific Zscore Distributions for CT and AD Individuals across Glycerophospholipid metabolism :
library(ggplot2) library(ggridges) library(reshape2)
library(ggbreak)
# Select pathway-specific (eg: hsa00564) Zscore from all four groups # Find the maximum length of the vectors max_length <- max( length(final_results_AD_female$hsa00564_Zscore), length(final_results_AD_male$hsa00564_Zscore), length(final_results_AsymAD_female$hsa00564_Zscore), length(final_results_AsymAD_male$hsa00564_Zscore), length(final_results_CT_female$hsa00564_Zscore), length(final_results_CT_male$hsa00564_Zscore) ) # Function to pad a vector with NA values pad_vector <- function(vec, length_out) { c(vec, rep(NA, length_out - length(vec))) } # Pad each vector to the maximum length hsa00564_AD_female_padded <- pad_vector(final_results_AD_female$hsa00564_Zscore, max_length) hsa00564_AD_male_padded <- pad_vector(final_results_AD_male$hsa00564_Zscore, max_length) hsa00564_AsymAD_female_padded <- pad_vector(final_results_AsymAD_female$hsa00564_Zscore, max_length) hsa00564_AsymAD_male_padded <- pad_vector(final_results_AsymAD_male$hsa00564_Zscore, max_length) hsa00564_CT_female_padded <- pad_vector(final_results_CT_female$hsa00564_Zscore, max_length) hsa00564_CT_male_padded <- pad_vector(final_results_CT_male$hsa00564_Zscore, max_length) # Combine into a single data frame hsa00564_Zscore_summary <- data.frame( hsa00564_AD_female = hsa00564_AD_female_padded, hsa00564_AD_male = hsa00564_AD_male_padded, hsa00564_AsymAD_female = hsa00564_AsymAD_female_padded, hsa00564_AsymAD_male = hsa00564_AsymAD_male_padded, hsa00564_CT_female = hsa00564_CT_female_padded, hsa00564_CT_male = hsa00564_CT_male_padded ) # View the combined data frame #head(hsa00564_Zscore_summary) # Print the table using DT datatable(hsa00564_Zscore_summary, options = list(pageLength = 5, autoWidth = TRUE))
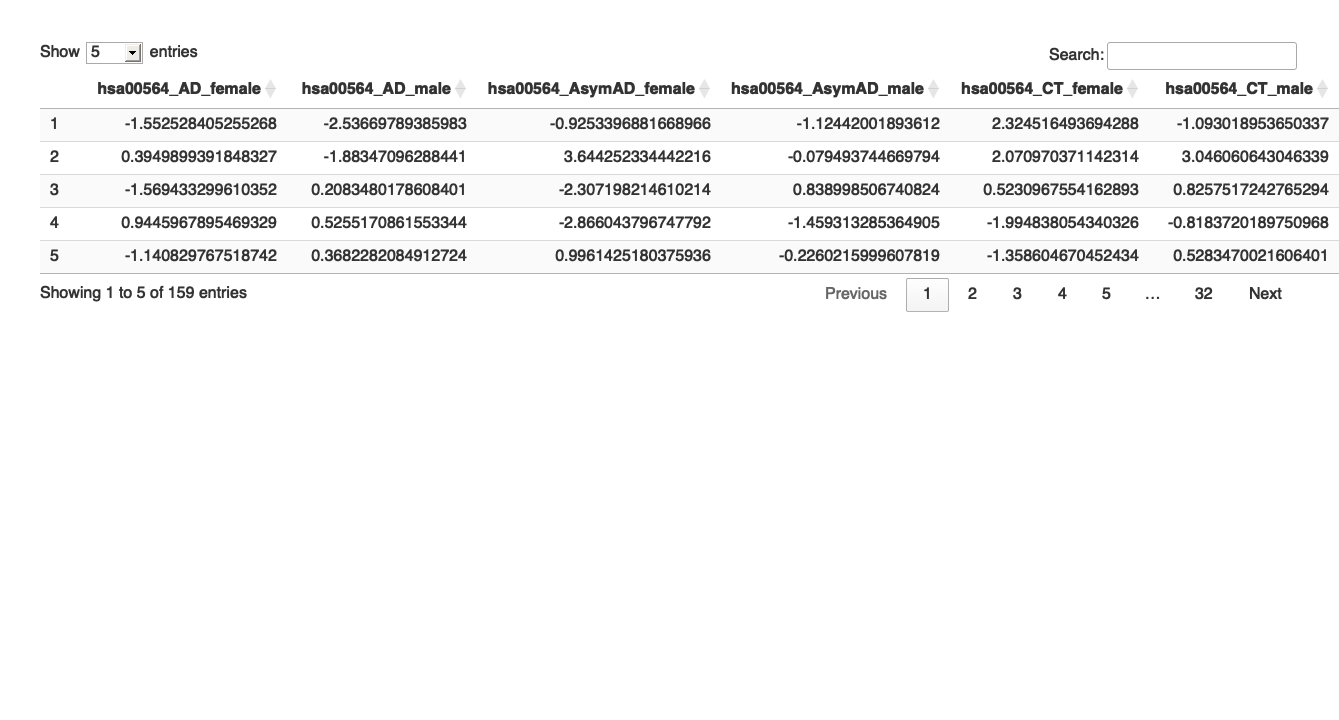
#write.table(hsa00564_Zscore_summary, "/Users/poddea/Desktop/hsa00564_Zscore_summary.txt", sep = "\t", quote = FALSE, row.names = FALSE) # Transform the data to long format hsa00564_Zscore_summary_long <- melt(hsa00564_Zscore_summary, variable.name = "Condition", value.name = "Value")
# Remove NA values (if any) hsa00564_Zscore_summary_long <- hsa00564_Zscore_summary_long[complete.cases(hsa00564_Zscore_summary_long), ] # Create the Ridgeline Plot: ggplot(hsa00564_Zscore_summary_long, aes(x = Value, y = Condition, fill = Condition)) + geom_density_ridges(scale = 1) + theme_ridges() + theme(legend.position = "none") + labs(title = "Comparison of Pathway-specific Zscore Distributions for CT, AD and AsymAD Individuals across Glycerophospholipid metabolism ", x = "Value", y = "Condition") + scale_fill_manual(values = c("tomato", "blue", "coral", "cornflowerblue", "pink", "lightblue"))
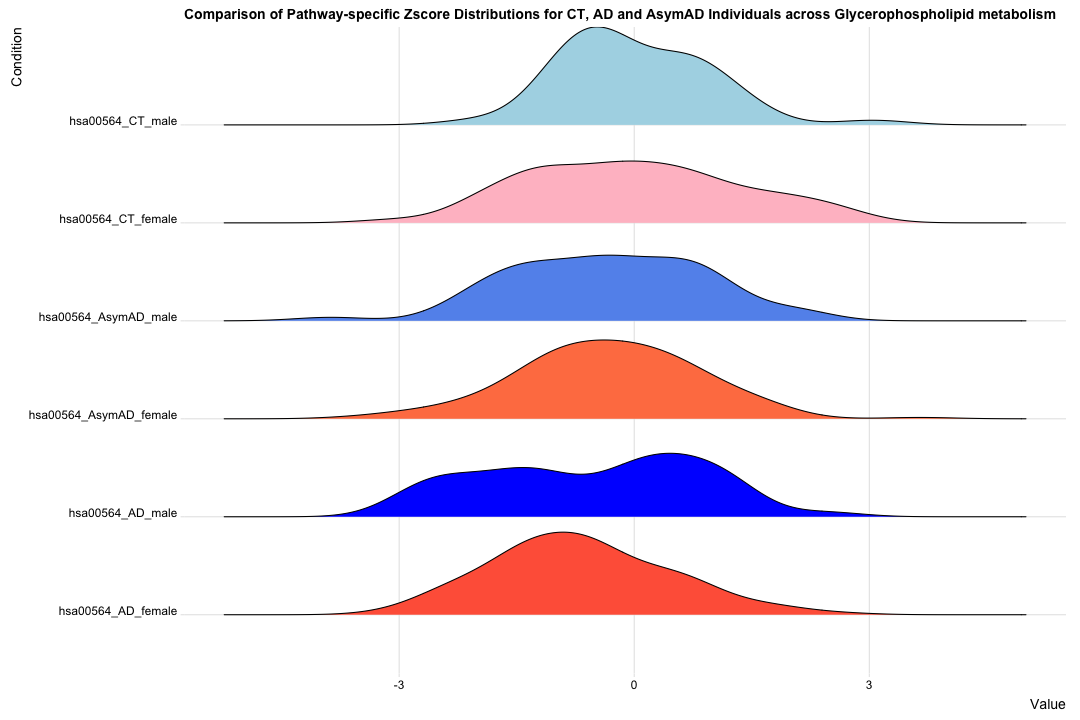
############################################################################################################################################################## # Filter the data for Female_CT and Female_AD data_filtered_AD_mf <- subset(hsa00564_Zscore_summary_long, Condition %in% c("hsa00564_AD_female", "hsa00564_AD_male")) data_filtered_CTAD_m <- subset(hsa00564_Zscore_summary_long, Condition %in% c("hsa00564_CT_male", "hsa00564_AD_male")) data_filtered_CTAD_f <- subset(hsa00564_Zscore_summary_long, Condition %in% c("hsa00564_CT_female", "hsa00564_AD_female")) # Create the overlapping density plot ggplot(data_filtered_AD_mf, aes(x = Value, fill = Condition)) + geom_density(alpha = 0.5) + # Set transparency for overlapping densities theme_minimal() + labs(title = "Density Plot for AD_male and AD_female across Glycerophospholipid metabolism pathway", x = "Z score", y = "Density") + scale_fill_manual(values = c("hsa00564_AD_female" = "red", "hsa00564_AD_male" = "blue")) + theme_minimal(base_size = 14) + theme( text = element_text(size = 20), legend.position = "right" )
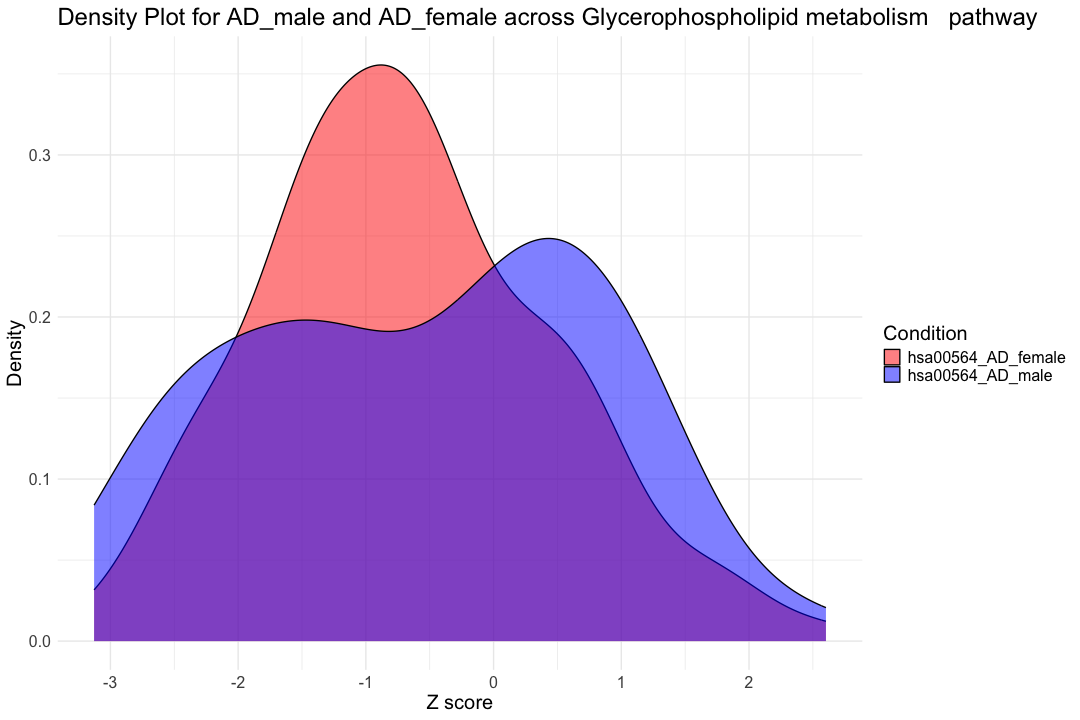
result_AD_mf <- wilcox.test(hsa00564_Zscore_summary$hsa00564_AD_male, hsa00564_Zscore_summary$hsa00564_AD_female, paired = TRUE, na.rm = TRUE) print(result_AD_mf)
## ## Wilcoxon signed rank exact test ## ## data: hsa00564_Zscore_summary$hsa00564_AD_male and hsa00564_Zscore_summary$hsa00564_AD_female ## V = 669, p-value = 0.2715 ## alternative hypothesis: true location shift is not equal to 0
ggplot(data_filtered_CTAD_m, aes(x = Value, fill = Condition)) + geom_density(alpha = 0.5) + # Set transparency for overlapping densities theme_minimal() + labs(title = "Density Plot for CT_male and AD_male across Glycerophospholipid metabolism pathway", x = "Z score", y = "Density") + scale_fill_manual(values = c("hsa00564_CT_male" = "lightblue", "hsa00564_AD_male" = "blue")) + theme(legend.title = element_blank())
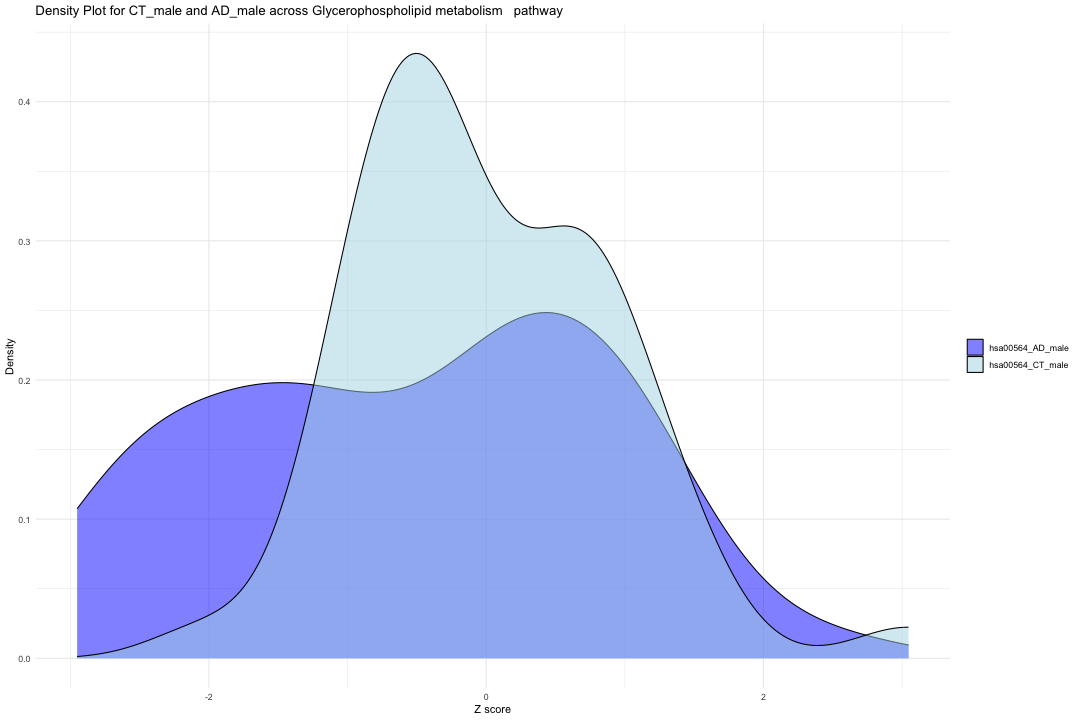
result_CTAD_m <- wilcox.test(hsa00564_Zscore_summary$hsa00564_CT_male, hsa00564_Zscore_summary$hsa00564_AD_male, paired = TRUE, na.rm = TRUE) print(result_CTAD_m)
## ## Wilcoxon signed rank exact test ## ## data: hsa00564_Zscore_summary$hsa00564_CT_male and hsa00564_Zscore_summary$hsa00564_AD_male ## V = 757, p-value = 0.01727 ## alternative hypothesis: true location shift is not equal to 0
ggplot(data_filtered_CTAD_f, aes(x = Value, fill = Condition)) + geom_density(alpha = 0.5) + # Set transparency for overlapping densities theme_minimal() + labs(title = "Density Plot for CT_female and AD_female across Glycerophospholipid metabolism ", x = "Z score", y = "Density") + scale_fill_manual(values = c("hsa00564_CT_female" = "pink", "hsa00564_AD_female" = "red")) + theme(legend.title = element_blank())
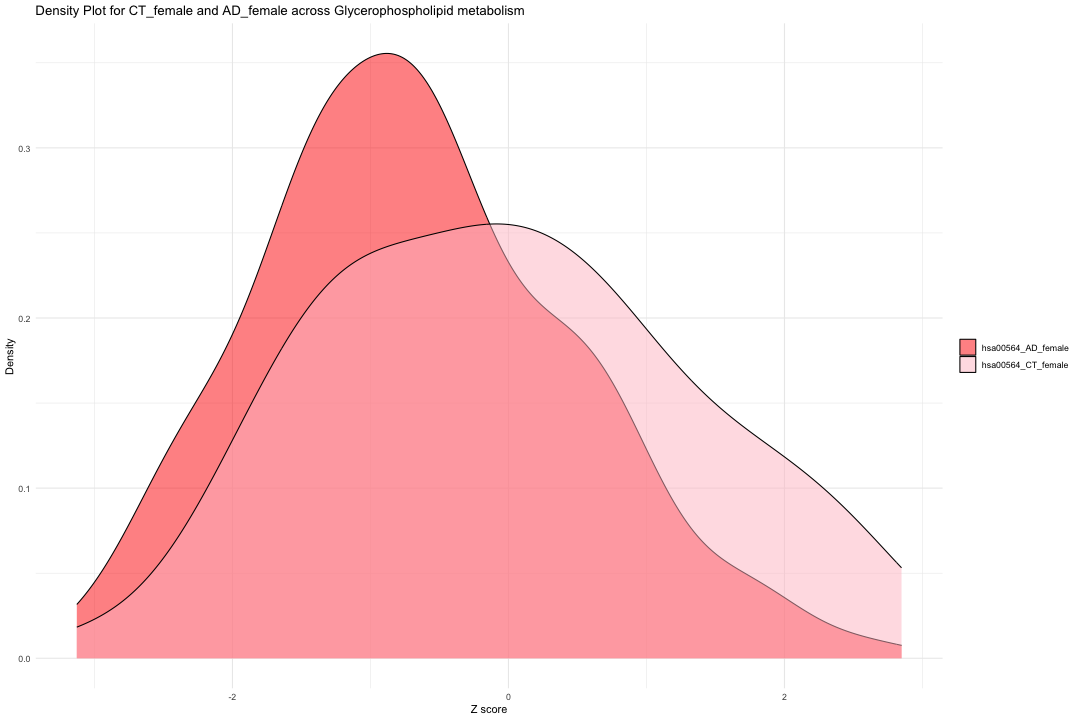
result_CTAD_f <- wilcox.test(hsa00564_Zscore_summary$hsa00564_CT_female, hsa00564_Zscore_summary$hsa00564_AD_female, paired = TRUE, na.rm = TRUE) print(result_CTAD_f)
## ## Wilcoxon signed rank test with continuity correction ## ## data: hsa00564_Zscore_summary$hsa00564_CT_female and hsa00564_Zscore_summary$hsa00564_AD_female ## V = 1631, p-value = 0.005182 ## alternative hypothesis: true location shift is not equal to 0
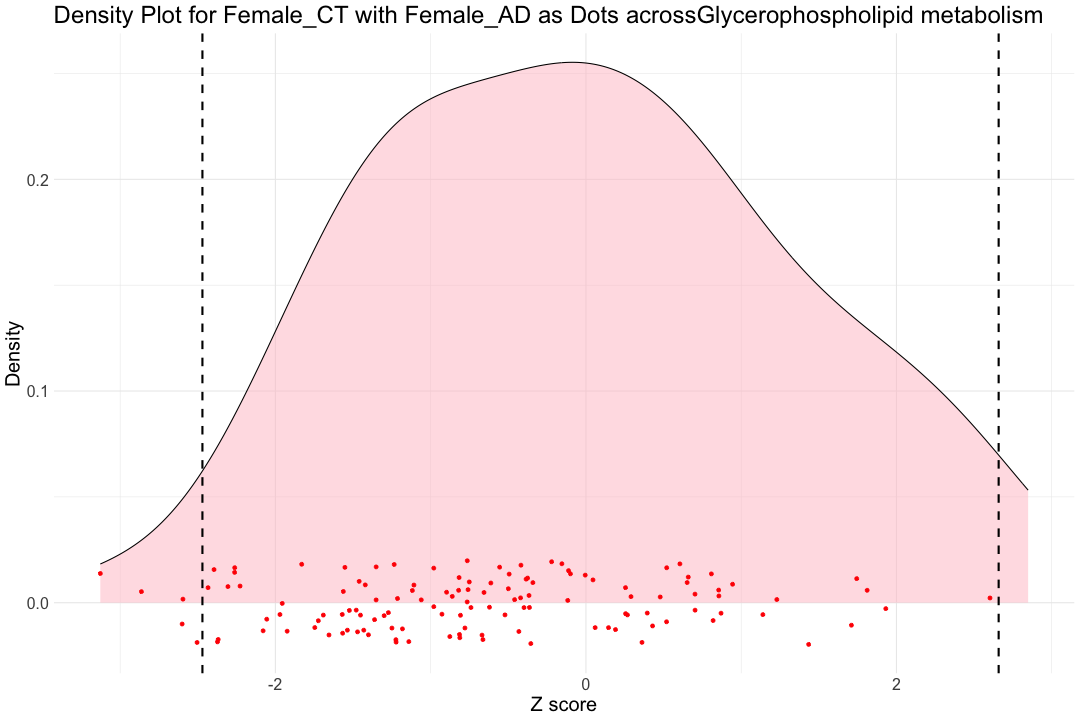
######################################################################################################################################################################## # Calculate the 1st and 99th percentiles of pathway-specific Z scores for the hsa00564 pathway within the Control individuals hsa00564_CT_female_1st <- quantile(hsa00564_Zscore_summary$hsa00564_CT_female, 0.01, na.rm = TRUE) hsa00564_CT_female_99th <- quantile(hsa00564_Zscore_summary$hsa00564_CT_female, 0.99, na.rm = TRUE) hsa00564_CT_male_1st <- quantile(hsa00564_Zscore_summary$hsa00564_CT_male, 0.01, na.rm = TRUE) hsa00564_CT_male_99th <- quantile(hsa00564_Zscore_summary$hsa00564_CT_male, 0.99, na.rm = TRUE) ggplot(hsa00564_Zscore_summary, aes(x = hsa00564_CT_female)) + geom_density(fill = "pink", alpha = 0.5) + # Density plot for Female_CT geom_point(aes(x = hsa00564_AD_female, y = 0), position = position_jitter(height = 0.02), color = "red") + # Dots for Female_AD labs(title = "Density Plot for Female_CT with Female_AD as Dots acrossGlycerophospholipid metabolism ", x = "Z score", y = "Density") + theme_minimal() + geom_vline(xintercept = hsa00564_CT_female_1st, color = "black", linetype = "dashed", size = 1) + geom_vline(xintercept = hsa00564_CT_female_99th, color = "black", linetype = "dashed", size = 1) + theme(text = element_text(size=20))
## Warning: Removed 91 rows containing non-finite outside the scale range ## (`stat_density()`).
## Warning: Removed 32 rows containing missing values or values outside the scale range ## (`geom_point()`).
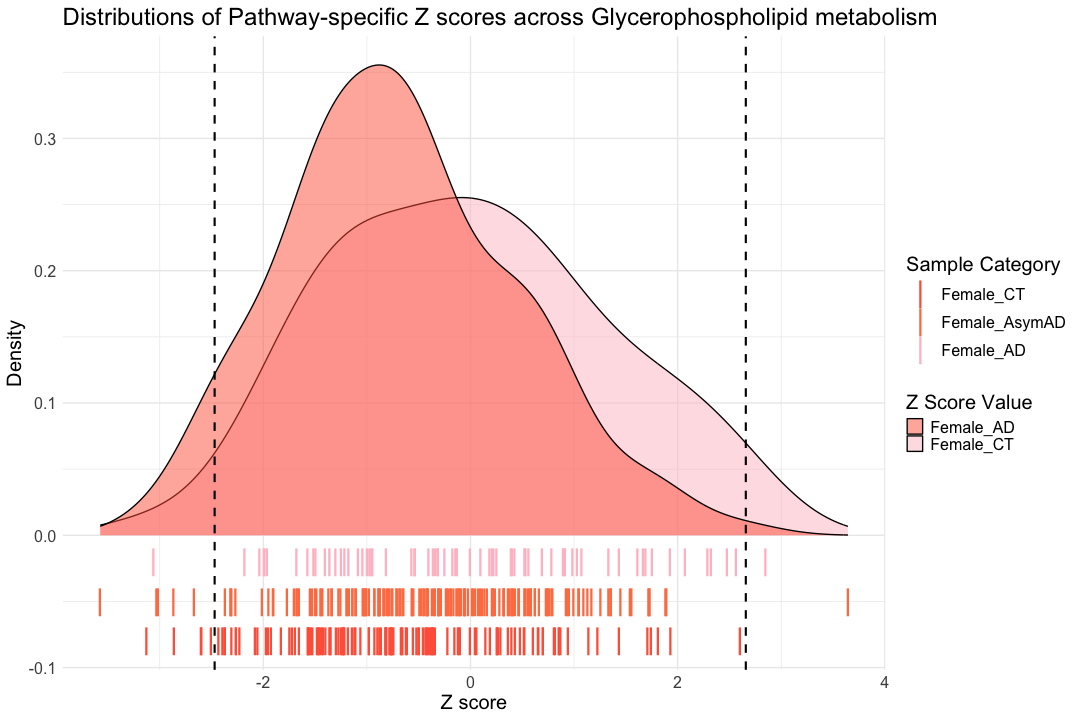
############################################################################################################################### ggplot() + # Density plot for Female_CT geom_density( data = hsa00564_Zscore_summary, aes(x = hsa00564_CT_female, fill = "Female_CT"), alpha = 0.5 ) + # Density plot for Female_AD geom_density( data = hsa00564_Zscore_summary, aes(x = hsa00564_AD_female, fill = "Female_AD"), alpha = 0.5 ) + # Mimic rug plot for Female_CT geom_point( data = hsa00564_Zscore_summary, aes(x = hsa00564_CT_female, y = -0.02, color = "Female_CT"), size = 10, shape = "|" ) + # Mimic rug plot for Female_AsymAD geom_point( data = hsa00564_Zscore_summary, aes(x = hsa00564_AsymAD_female, y = -0.05, color = "Female_AsymAD"), size = 10, shape = "|" ) + # Mimic rug plot for Female_AD geom_point( data = hsa00564_Zscore_summary, aes(x = hsa00564_AD_female, y = -0.08, color = "Female_AD"), size = 10, shape = "|" ) + # Labels and themes labs( title = "Distributions of Pathway-specific Z scores across Glycerophospholipid metabolism ", x = "Z score", y = "Density", fill = "Z Score Value", color = "Sample Category" ) + theme_minimal(base_size = 14) + theme( text = element_text(size = 20), legend.position = "right" ) + # Vertical dashed lines for Female_CT geom_vline(xintercept = hsa00564_CT_female_1st, color = "black", linetype = "dashed", size = 1) + geom_vline(xintercept = hsa00564_CT_female_99th, color = "black", linetype = "dashed", size = 1) + # Manually specify the color scales for the legends scale_fill_manual( values = c("Female_CT" = "pink", "Female_AD" = "tomato"), labels = c("Female_AD", "Female_CT") ) + scale_color_manual( values = c("Female_CT" = "pink","Female_AsymAD" = "coral", "Female_AD" = "tomato"), labels = c("Female_CT", "Female_AsymAD", "Female_AD") ) #+
## Warning: Removed 91 rows containing non-finite outside the scale range ## (`stat_density()`).
## Warning: Removed 32 rows containing non-finite outside the scale range ## (`stat_density()`).
## Warning: Removed 91 rows containing missing values or values outside the scale range ## (`geom_point()`).
## Warning: Removed 32 rows containing missing values or values outside the scale range ## (`geom_point()`).
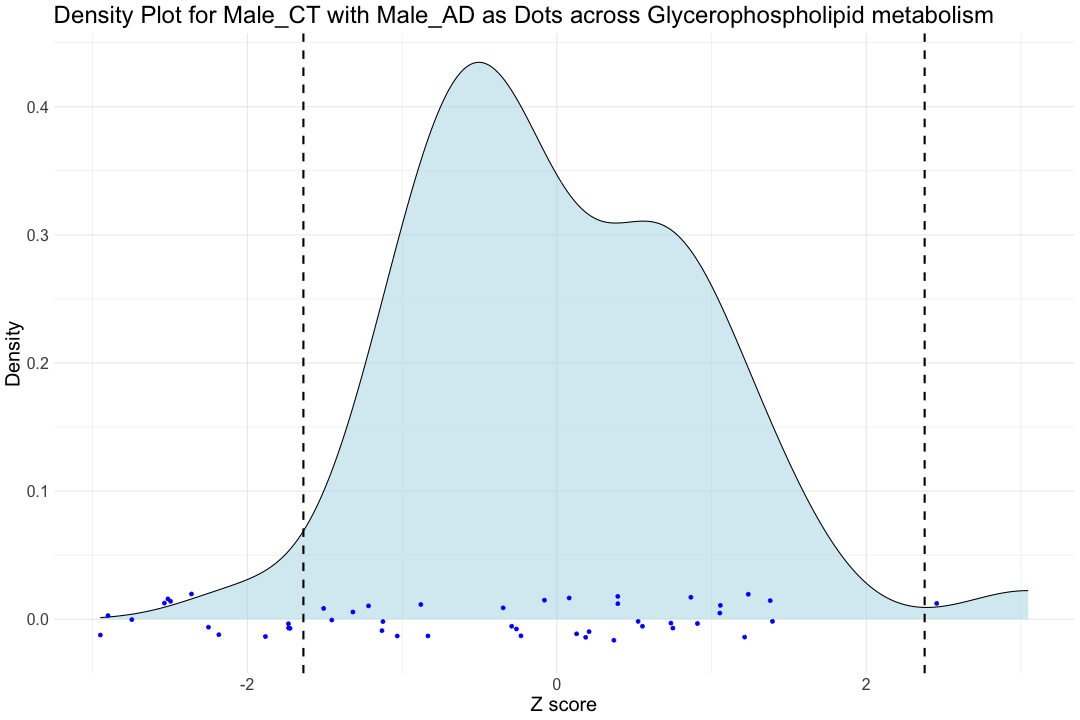
# Break the X axis manually to make the plot more visible #scale_x_break(c(7.5, 17.5), scales = 0.3, ticklabels = c(7.5, 17.5)) ############################################################################################################################### ggplot(hsa00564_Zscore_summary, aes(x = hsa00564_CT_male)) + geom_density(fill = "lightblue", alpha = 0.5) + # Density plot for Female_CT geom_point(aes(x = hsa00564_AD_male, y = 0), position = position_jitter(height = 0.02), color = "blue") + # Dots for Male_AD labs(title = "Density Plot for Male_CT with Male_AD as Dots across Glycerophospholipid metabolism ", x = "Z score", y = "Density") + theme_minimal() + geom_vline(xintercept = hsa00564_CT_male_1st, color = "black", linetype = "dashed", size = 1) + geom_vline(xintercept = hsa00564_CT_male_99th, color = "black", linetype = "dashed", size = 1) + theme(text = element_text(size=20))
## Warning: Removed 113 rows containing non-finite outside the scale range ## (`stat_density()`).
## Warning: Removed 112 rows containing missing values or values outside the scale range ## (`geom_point()`).
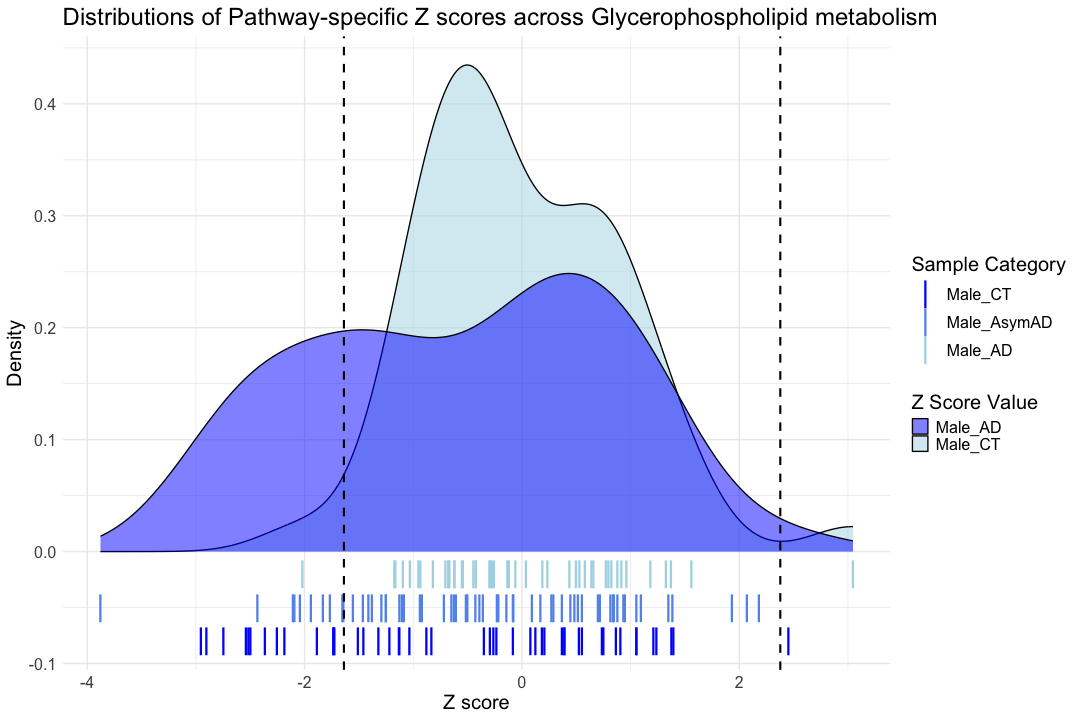
############################################################################################################################### ggplot() + # Density plot for Male_CT geom_density( data = hsa00564_Zscore_summary, aes(x = hsa00564_CT_male, fill = "Male_CT"), alpha = 0.5 ) + # Density plot for Male_AD geom_density( data = hsa00564_Zscore_summary, aes(x = hsa00564_AD_male, fill = "Male_AD"), alpha = 0.5 ) + # Mimic rug plot for Male_CT geom_point( data = hsa00564_Zscore_summary, aes(x = hsa00564_CT_male, y = -0.02, color = "Male_CT"), size = 10, shape = "|" ) + # Mimic rug plot for Male_AsymAD geom_point( data = hsa00564_Zscore_summary, aes(x = hsa00564_AsymAD_male, y = -0.05, color = "Male_AsymAD"), size = 10, shape = "|" ) + # Mimic rug plot for Male_AD geom_point( data = hsa00564_Zscore_summary, aes(x = hsa00564_AD_male, y = -0.08, color = "Male_AD"), size = 10, shape = "|" ) + # Labels and themes labs( title = "Distributions of Pathway-specific Z scores across Glycerophospholipid metabolism ", x = "Z score", y = "Density", fill = "Z Score Value", color = "Sample Category" ) + theme_minimal(base_size = 14) + theme( text = element_text(size = 20), legend.position = "right" ) + # Vertical dashed lines for Male_CT geom_vline(xintercept = hsa00564_CT_male_1st, color = "black", linetype = "dashed", size = 1) + geom_vline(xintercept = hsa00564_CT_male_99th, color = "black", linetype = "dashed", size = 1) + # Manually specify the color scales for the legends scale_fill_manual( values = c("Male_CT" = "lightblue", "Male_AD" = "blue"), labels = c("Male_AD", "Male_CT") ) + scale_color_manual( values = c("Male_CT" = "lightblue", "Male_AsymAD" = "cornflowerblue", "Male_AD" = "blue"), labels = c("Male_CT", "Male_AsymAD", "Male_AD") ) #+
## Warning: Removed 113 rows containing non-finite outside the scale range ## (`stat_density()`).
## Warning: Removed 112 rows containing non-finite outside the scale range ## (`stat_density()`).
## Warning: Removed 113 rows containing missing values or values outside the scale range ## (`geom_point()`).
## Warning: Removed 98 rows containing missing values or values outside the scale range ## (`geom_point()`).
## Warning: Removed 112 rows containing missing values or values outside the scale range ## (`geom_point()`).
# Break the X axis manually to make the plot more visible #scale_x_break(c(10, 20), scales = 0.3, ticklabels = c(10, 20))