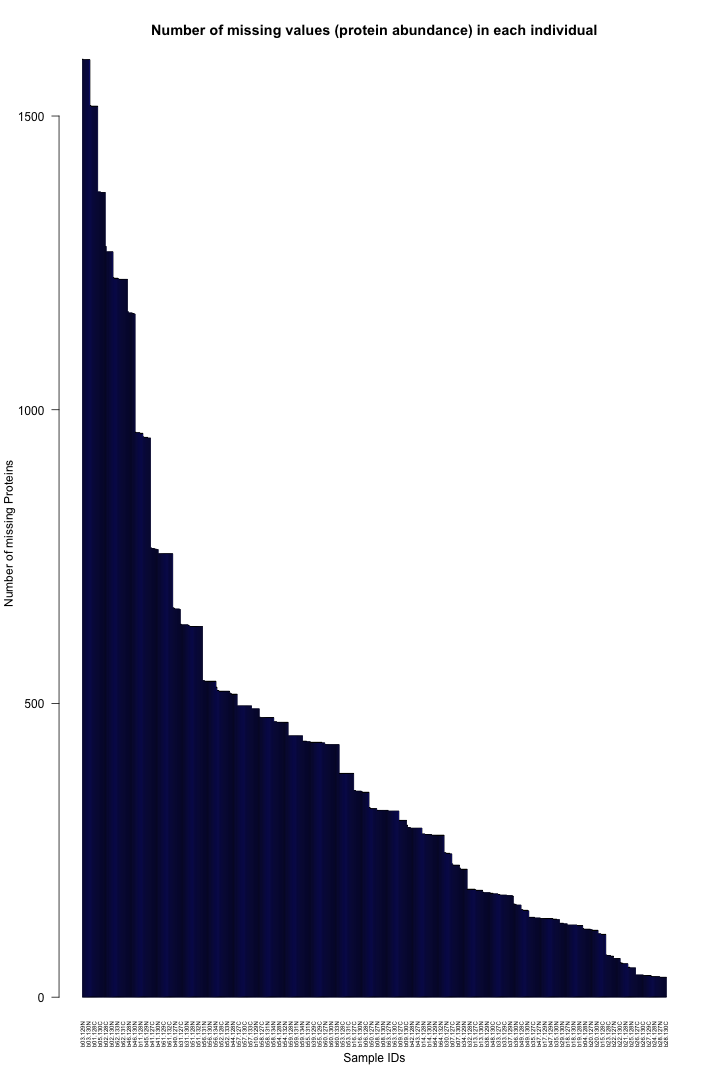
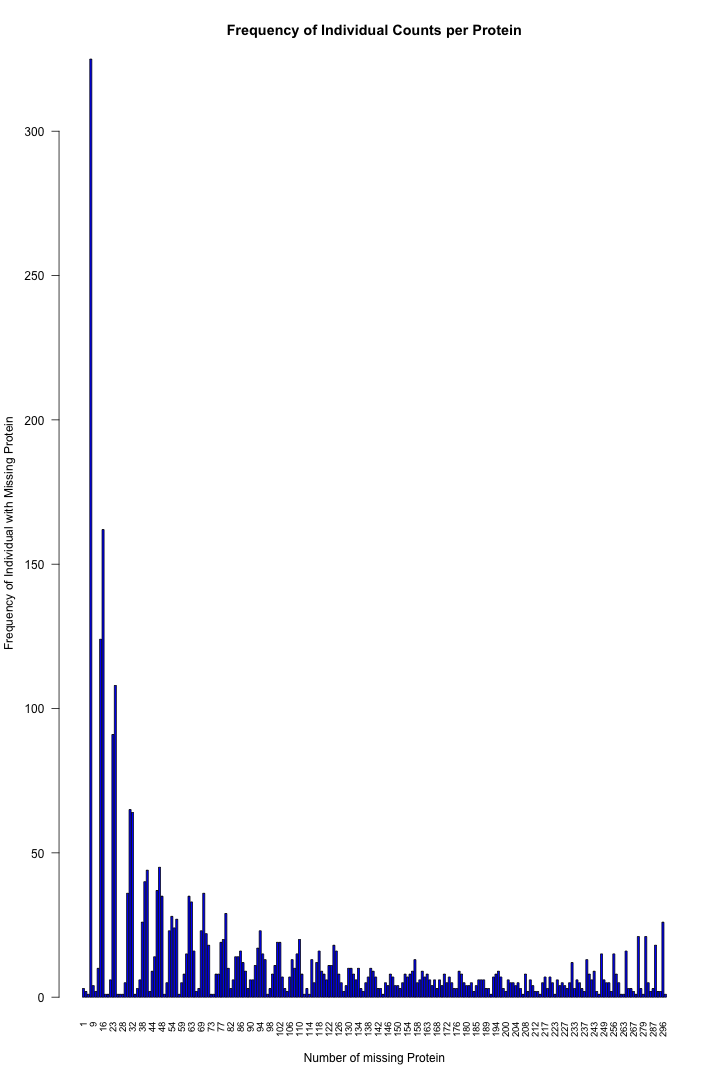
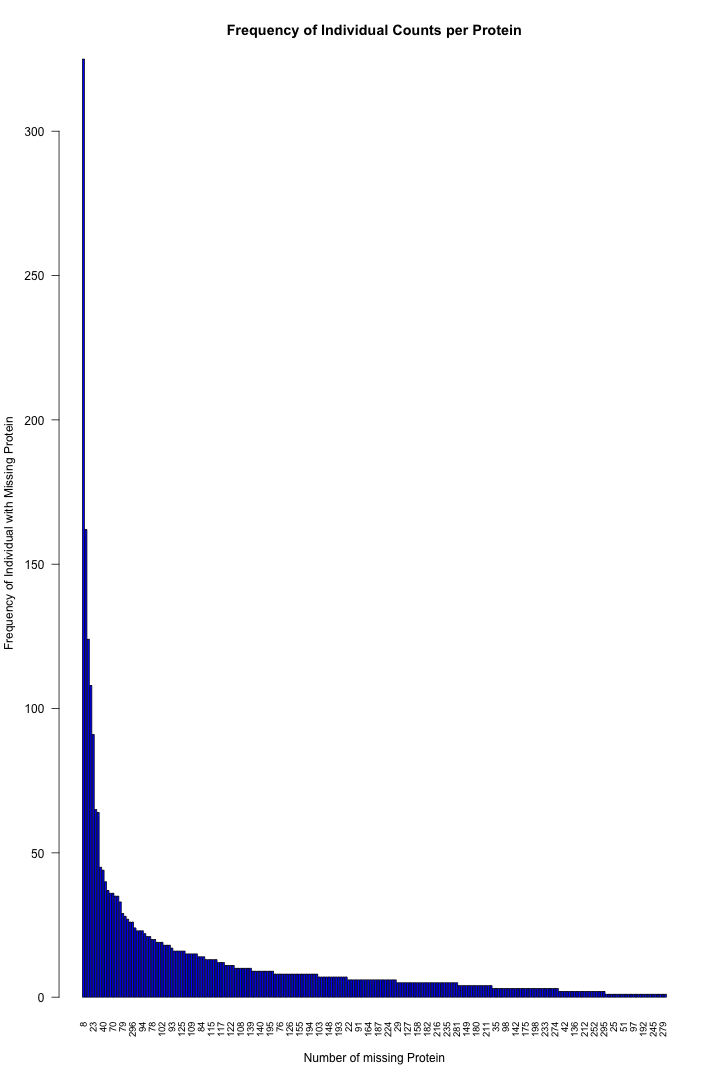
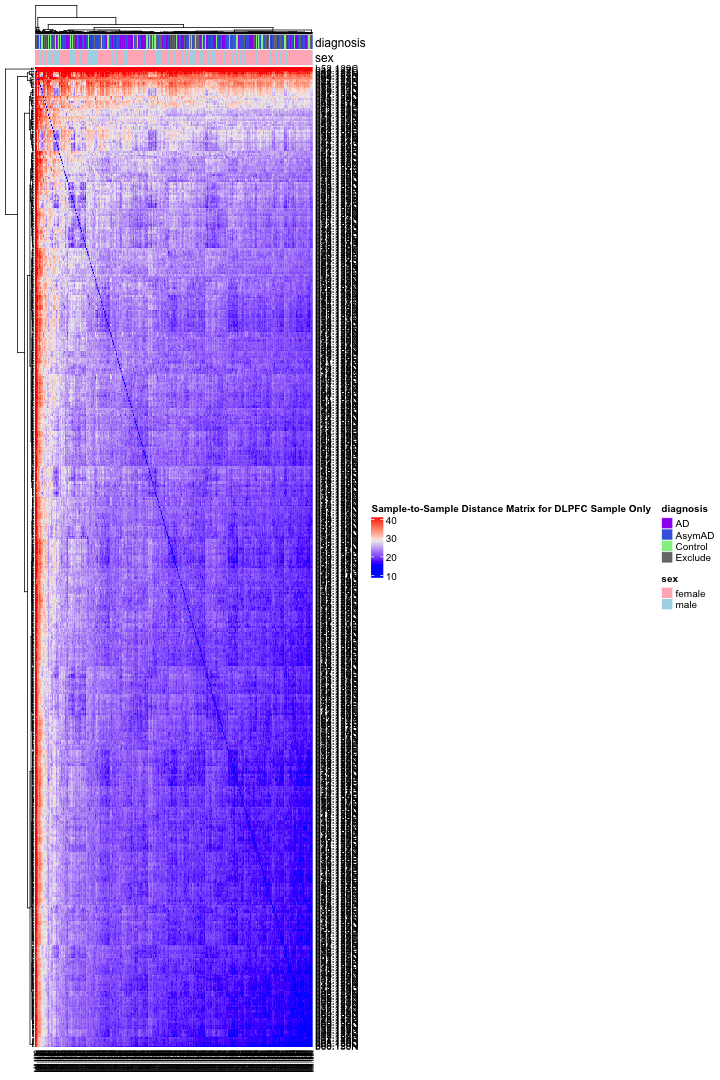
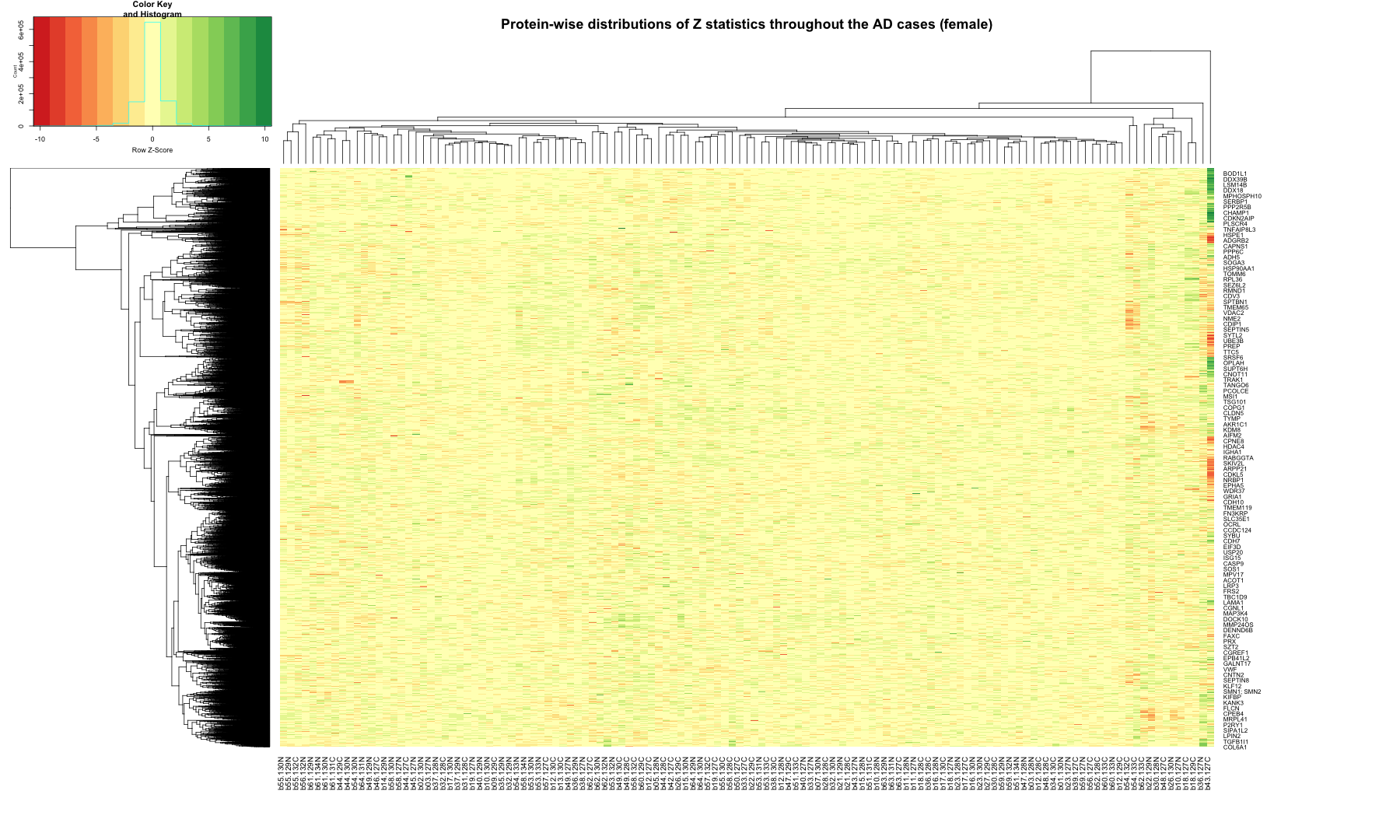
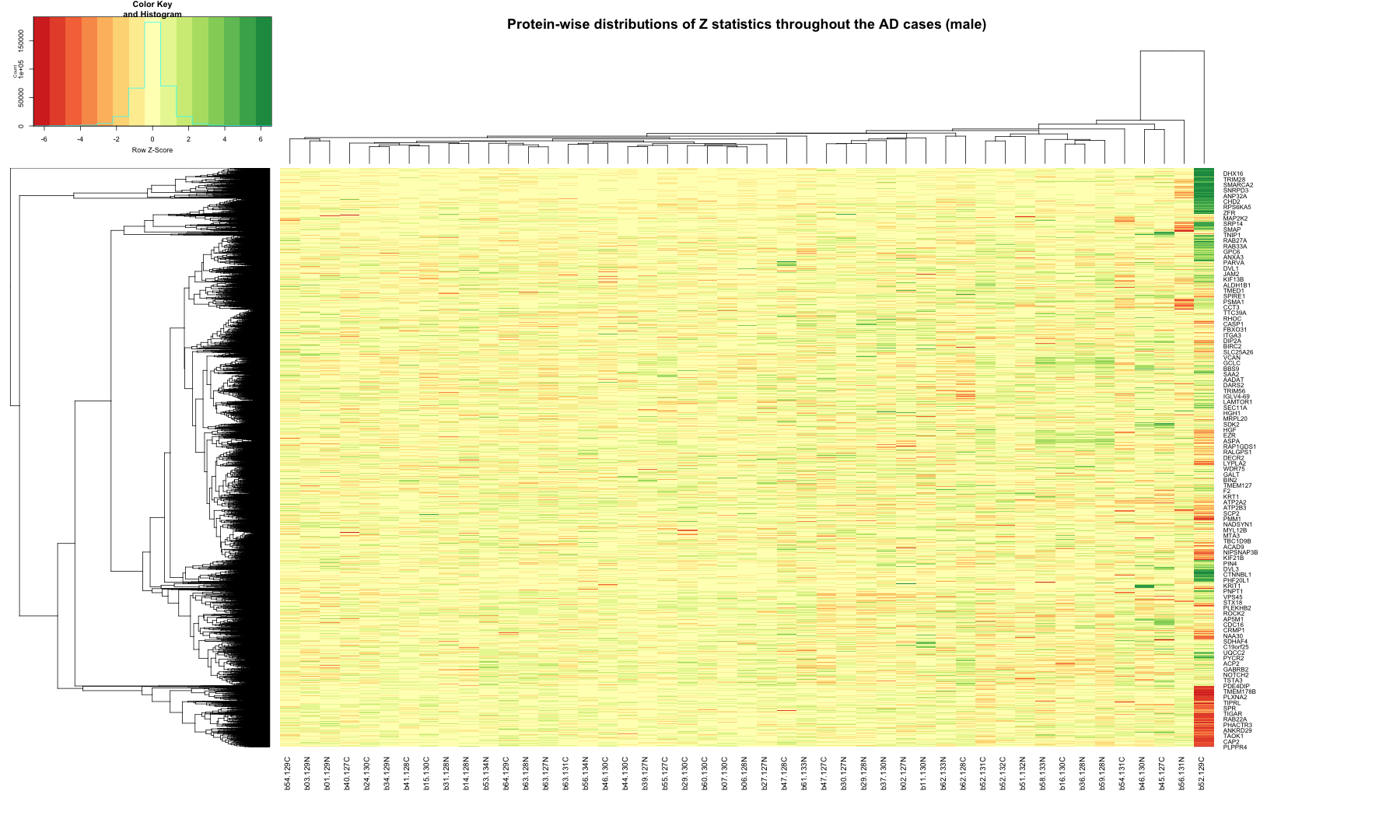
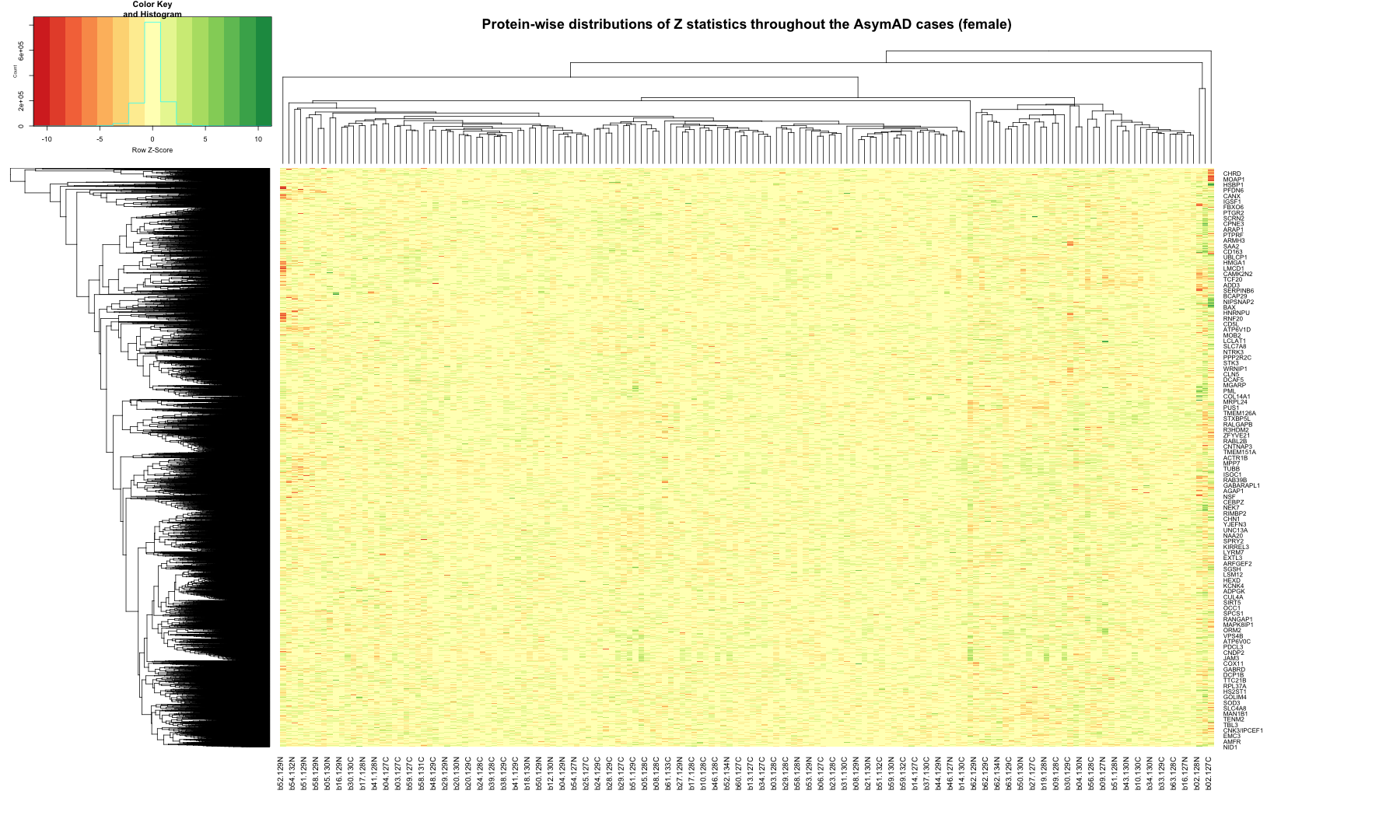
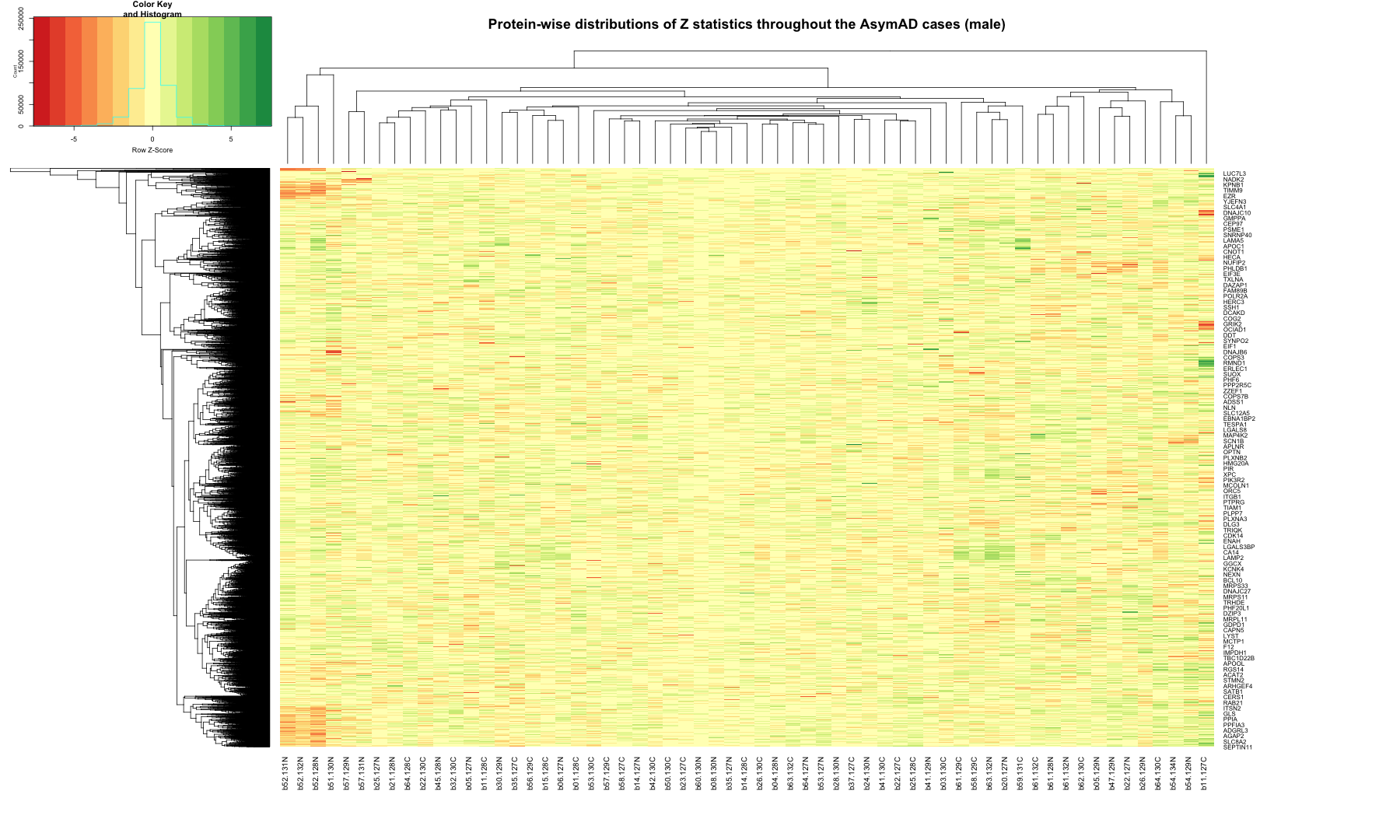
# Get the list of proteins from the protein_data_filtered
Protein_name <- protein_data_filtered$Protein_name
# Get the list of samples from the protein_data_filtered
common_samples <- colnames(protein_data_filtered)
# Select a sub-set of the sample (Tissue: DLPFC) from protein_metadata_ROSMAP file
sample_data_DLPFC <- filter(protein_metadata_ROSMAP, tissue == "dorsolateral prefrontal cortex")
# Create a subset of the DLPFC sample description file with control samples only
sample_data_DLPFC_contorl <- sample_data_DLPFC[grep("Control", sample_data_DLPFC$JohnsonDx), ]
dim(sample_data_DLPFC_contorl)
# Create a subset of the DLPFC sample description file with AD samples only
#sample_data_DLPFC_case <- sample_data_DLPFC[grep("AD", sample_data_DLPFC$JohnsonDx), ]
sample_data_DLPFC_case <- sample_data_DLPFC[sample_data_DLPFC$JohnsonDx == "AD", ]
dim(sample_data_DLPFC_case)
# Create a subset of the DLPFC sample description file with AsymAD samples only
sample_data_DLPFC_AsymAD <- sample_data_DLPFC[grep("AsymAD", sample_data_DLPFC$JohnsonDx), ]
dim(sample_data_DLPFC_AsymAD)
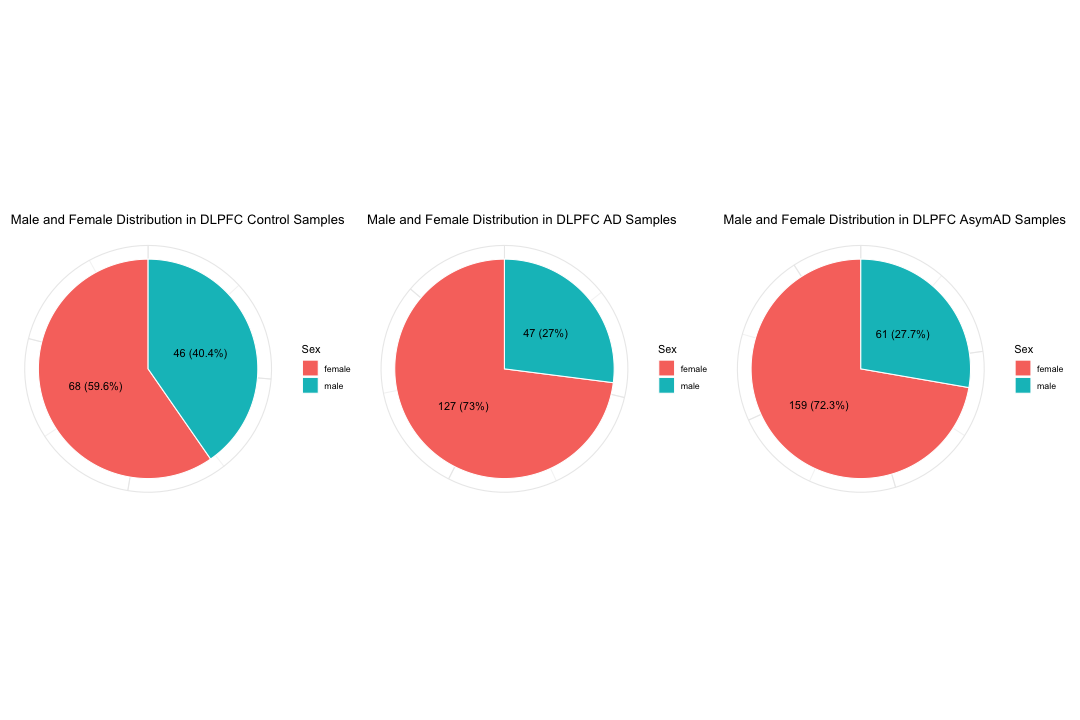
# Calculate the counts and percentages of each sex in control samples
sex_counts_control <- table(sample_data_DLPFC_contorl$sex)
sex_percentages_control <- prop.table(sex_counts_control) * 100
# Create a pie chart to show the distribution of Male and Female ratio within the control samples
pie_data_control <- data.frame(sex = names(sex_counts_control), count = as.numeric(sex_counts_control), percentage = sex_percentages_control)
P1 <- ggplot(pie_data_control, aes(x = "", y = count, fill = sex)) +
geom_bar(stat = "identity", width = 1, color = "white") +
coord_polar("y") +
geom_text(aes(label = paste0(count, " (", round(sex_percentages_control, 1), "%)")), position = position_stack(vjust = 0.5)) + # Add text labels with percentages
labs(title = "Male and Female Distribution in DLPFC Control Samples",
fill = "Sex") +
theme_minimal() +
theme(axis.text = element_blank(),
axis.title = element_blank(),
legend.position = "right")
# Calculate the counts and percentages of each sex in AD samples
sex_counts_case <- table(sample_data_DLPFC_case$sex)
sex_percentages_case <- prop.table(sex_counts_case) * 100
# Create a pie chart to show the distribution of Male and Female ratio within the AD samples
pie_data_case <- data.frame(sex = names(sex_counts_case), count = as.numeric(sex_counts_case), percentage = sex_percentages_case)
P2 <- ggplot(pie_data_case, aes(x = "", y = count, fill = sex)) +
geom_bar(stat = "identity", width = 1, color = "white") +
coord_polar("y") +
geom_text(aes(label = paste0(count, " (", round(sex_percentages_case, 1), "%)")), position = position_stack(vjust = 0.5)) + # Add text labels with percentages
labs(title = "Male and Female Distribution in DLPFC AD Samples",
fill = "Sex") +
theme_minimal() +
theme(axis.text = element_blank(),
axis.title = element_blank(),
legend.position = "right")
# Calculate the counts and percentages of each sex in AsymAD samples
sex_counts_AsymAD <- table(sample_data_DLPFC_AsymAD$sex)
sex_percentages_AsymAD <- prop.table(sex_counts_AsymAD) * 100
# Create a pie chart to show the distribution of Male and Female ratio within the AsymAD samples
pie_data_AsymAD <- data.frame(sex = names(sex_counts_AsymAD), count = as.numeric(sex_counts_AsymAD), percentage = sex_percentages_AsymAD)
P3 <- ggplot(pie_data_AsymAD, aes(x = "", y = count, fill = sex)) +
geom_bar(stat = "identity", width = 1, color = "white") +
coord_polar("y") +
geom_text(aes(label = paste0(count, " (", round(sex_percentages_AsymAD, 1), "%)")), position = position_stack(vjust = 0.5)) + # Add text labels with percentages
labs(title = "Male and Female Distribution in DLPFC AsymAD Samples",
fill = "Sex") +
theme_minimal() +
theme(axis.text = element_blank(),
axis.title = element_blank(),
legend.position = "right")
library(patchwork)
P1 | P2 | P3